Manjak 21–hidroksilaze (CYP21A2) uzrokuje poremećenu pretvorbu steroidnih prekursora u kortizol, u nekih slučajeva i u aldosteron, dovodeći do virilizacije te ponekad teške hiponatrijemije i hiperkalijemije. Akumulirani prekursori hormona usmjereni su u sintezu androgena što uzrokuje virilizaciju. Dijagnoza se postavlja određivanjem kortizola, njegovih prekursora i androgena nadbubrežne žlijezde, ponekad nakon primjene ACTH. Liječi se glukokortikoidom uz, ako je potrebno, mineralokortikoid, te u nekih bolesnica s dvosmislenim spolovilom, kirurškom rekonstrukcijom.
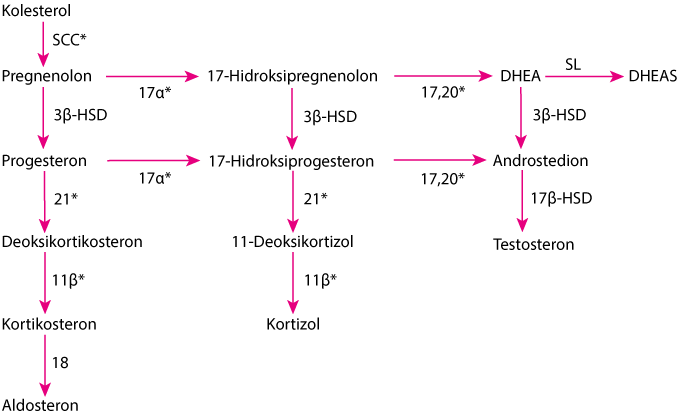
Nedostatak 21–hidroksilaze uzrokuje 90% svih slučajeva kongenitalne adrenalne hiperplazije. Incidencija iznosi od 1/10.000 do 1/15.000 živorođene djece. Težina bolesti ovisi o specifičnoj mutaciji CYP21A2 i stupnju nedostatka enzima. Nedostatak u cijelosti ili djelomično blokira pretvaranje 17-hidroksiprogesterona u 11-deoksikortizol, prekursora kortizola i pretvorbu progesterona u deoksikortikosteron, prekursora aldosterona. Budući da je sinteza kortizola smanjena, razina ACTH raste što uzrokuje nakupljanje prekursora kortizola (npr. 17-hidroksiprogesterona) i prekomjerne proizvodnje androgena nadbubrežne žlijezde dehidroepiandrosterona (DHEA) i androstendiona. Deficit aldosterona može dovesti do gubitka soli, hiponatremija, i hiperklemija (1, 2).
Sinteza hormona nadbubrežne žlijezde
 |
|
Adrenokortikotropni hormon (ACTH)
|
|
11β = 11β-hidroksilaza (P-450c11); 17α = 17α-hidroksilaza (P-450c17); 17,20 = 17,20 liaza (P-450c17); 18 = aldosteron sintetaza (P-450aldo); 21 = 21-hidroksilaza (P-450c21); DHEA = dehidroepiandrosteron; DHEAS = DHEA sulfat; 3β-HSD = 3β-hidroksisteroid dehidrogenaza (3β2-HSD); 17β-HSD = 17β-hidroksisteroid dehidrogenaza (17β-HSD); SCC = cijepanje bočnog lanca (P-450scc); SL = sulfotransferaza (SULT1A1, SULT1E1).
|
Klasični manjak 21-hidroksilaze
Klasični manjak 21-hidroksilaze može biti podijeljena u 2 oblika:
U oba oblika, razina androgena je povišena, što uzrokuje virilizaciju.
Oblik s gubitkom soli je najteži i čini 70% slučajeva klasičnog nedostatka 21-hidrolaze; postoji potpuni nedostatak enzimske aktivnosti što dovodi do vrlo niskih razina kortizola i aldosterona. Zbog toga što uopće nema izlučivanja aldosterona, dolazi do gubitka soli, što dovodi do hiponatrijemije, hiperkalijemije i povećane aktivnosti renina u plazmi.
Kod jednostavnog virilizirajućeg oblika, sinteza kortizol je umanjena, što dovodi do povećane aktivnosti androgena, ali postoji dovoljna aktivnost enzima za održavanje normalne, ili tek neznatno smanjene, proizvodnje aldosterona.
Ne-klasični manjak 21-hidroksilaze
Ne-klasični manjak 21-hidroksilaze je češći od klasičnog manjka 21-hidroksilaze. Incidencija varira od 1/1000 do 1/2000 živorođenih u Bijeloj populaciji (0,1 do 0,2%) do 1-2% u određenim etničkim skupinama (npr. Aškenazi židova). Ne-klasični manjak 21-hidroksilaze izaziva blažu formu poremećaj u kojem se nalazi 20 do 50% aktivnosti 21-hidroksilaze (u odnosu na 0-5% aktivnosti u klasičnom manjku 21-hidroksilaze). Gubitak soli je odsutan zbog urednih razina aldosterona i kortizola, međutim, razinama androgena je blago povišena, što rezultira blagim suviškom androgena u djetinjstvu ili odrasloj dobi.
Literatura
-
1. Witchel SF: Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol 30(5):520–534, 2017. doi: 10.1016/j.jpag.2017.04.001
-
2. El-Maouche D, Arlt W, Merke DP: Congenital adrenal hyperplasia. Lancet 390(10108):2194–2210, 2017. doi: 10.1016/S0140-6736(17)31431-9
Simptomi i znakovi
Oblik bolesti s gubitkom soli uzrokuje hiponatrijemiju (ponekad tešku), hiperkalijemiju, hipotenziju kao i virilizaciju. Ako se ne dijagnosticira i ne liječi, ovaj oblik može dovesti do po život opasne adrenalne krize, s povraćanjem, proljevom, hipoglikemijom, hipovolemijom i šokom.
Djevojčice s ovim oblikom bolesti imaju dvosmisleno vanjsko spolovilo, povećan klitoris, srasle velike usne i urogenitalni sinus umjesto odvojenih otvora mokraćne cijevi i rodnice. Muška djeca u pravilu pokazuju normalan razvoj spolovila, što može odgoditi dijagnozu gubioca soli; zahvaćeni dječaci često se otkriju samo kroz rutinski novorođenački probir. Osim ako nisu otkriveni novorođenačkim probirom, dječaci s jednostavnim virilizirajućim oblikom mogu biti dijagnosticirani tek za par godina, kada se razviju znakovi suviška androgena. Kad je nedostatak enzima puno blaži, novorođenčad pokazuje slabu ili nikakvu virilizaciju, no višak androgena se očituje kasnije, ranom pojavom pubične dlakavosti i ubrzanjem rasta u oba spola, povećanjem klitorisa u djevojčica te povećanjem penisa i ranijim produbljenjem glasa u dječaka.
Djeca s ne-klasičnim manjkom 21-hidroksilaze nemaju simptome pri rođenju i obično se ne prezentiraju do djetinjstva ili adolescencije. U ženske djece mogu imati rani razvoj stidne dlakavosti, ubrzanu koštanu dob, hirzutizam, oligomenoreju i/ili akne; ovi simptomi mogu sličiti sindromu policističnih jajnika. U muške djece mogu imati rani razvoj stidne dlakavosti, ubrzani rasta i ubrzanu koštanu dob.
U ženske djece, osobito one s oblikom bolesti praćenim gubitkom soli, u odrasloj dobi može biti poremećena reproduktivna sposobnost; mogu imati sraštene labije i anovulatorne cikluse ili amenoreju. Neka muška djeca, s oblikom praćenim gubitkom soli odrasloj dobi su fertilni, dok drugi mogu razviti testikularne tumore (benigne intratestikularne mase sastavljene od nadbubrežne tkiva koje hipertrofiju pod kroničnom stimulacijom ACTH), disfunkciju Leydigovih stanica, sniženu razinu testosterona, i poremećenu spermatogenezu. Većina pogođenih muškaraca s oblikom bolesti bez gubitka soli su fertilni, čak i ako nisu liječeni, no u nekih je spermatogeneza poremećena.
Dijagnoza
Rutinski novorođenački probir obično uključuje mjerenje serumske razine 17-hidroksiprogesterona. Ako je razina povišena, dijagnoza nedostatak 21-hidroksilaze potvrđuje se određivanjem niske razine kortizola u krvi i visokim razinama DHEA, androstendiona i testosterona u krvi. Dijagnoza je rijetko nesigurna, i tada se razina navedenih hormona određuje prije, i nakon 60 minuta primjene ACTH (ACTH ili kosintropinski stimulacijski test). Ukoliko to sugerira neki podatak iz obiteljske anamneze, potrebno je učiniti genetsko testiranje. U bolesnika koji simptome razviju kasnije, stimulacijski test s ACTH može pomoći, no može biti potrebna i genotipizacija.
U djece s oblikom praćenim s gubitkom soli imaju hiponatrijemiju i hiperkalemiju; niske razine deoksikortikosterona, kortikosterona i aldosterona; i visoke razine renina.
Prenatalni probir i dijagnoza (i eksperimentalni tretman) su moguće; CYP21 gen se analizira ako je rizik visok (npr. fetus ima brata ili sestru s genetskim defektom). U djece i odraslih se može utvrditi nosilaštvo (heterozigotnost).
Liječenje
-
Supstitucija kortikosteroidima
-
Supstitucija mineralokortikoidima (kod gubitka soli)
-
Ponekad rekonstruktivna kirurgija
Adrenalna kriza u dojenčkoj dobi može rezultirati hipotenzijom i šokom, praćenim hiponatrijemijom i hiperkalemijom. Ovisno o dobi, može se manifestirati letargijom, umorom, povraćanjem, smetenošću ili čak komom. Adrenalnu krizu u djece treba hitno liječiti nadoknadom IV tekućine. Stres doze hidrokortizon (100 mg/m2/dnevno) primjenjuju se kontinuirano IV infuzijom kako bi se spriječio nastanak adrenalne krize ako se sumnja na oblik s gubitkom soli; kroz nekoliko tjedna se smanjuje do fiziološke nadomjesne doze.
Liječenje održavanja klasični nedostatak 21-hidroksilaze is corticosteroids as replacement for deficient steroids (tje kortikosteroidi kao zamjena za deficitarne steroide (obično, oralni hidrokortizon u obliku tableta 3,5 do 5 mg/m2, s ukupnom dnevnom dozom obično ≤ 20 mg/m2). Za dojenčad i mlađu djecu, tablete se zdrobe ili razdvoje i pomiješaju s tekućinom. Granule s niskom dozom hidrokortizona sada su dostupne za liječenje dojenčadi s kongenitalnom adrenalnom hiperplazijom i mogu poboljšati točnost doziranja.
Hidrokortizon je poželjan kod djece koja rastu jer je manje potentan od drugih kortikosteroidnih pripravaka i uzrokuje manju supresiju rasta. Postpubertetski adolescenti i odrasli mogu se liječiti s prednizonom 5 do 7,5 mg po jednom/ dnevno ili 2,5 do 3,75 mg dva puta dnevno, ili deksametazonom 0,25 do 0,5 mg jednom/dnevno ili 0.125 do 0.25 mg.
Terapijski odgovor se u dojenčadi kontrolira svaka 3 mjeseca, a u djece >12 mjeseci svaka 3 do 4 mjeseca. Pretjerana primjena kortikosteroida dovodi do jatrogene Cushingove bolesti koja uzrokuje pretilost, usporeni rast i zakasnjelo sazrijevanje kostiju. Nedovoljna primjena dovodi do nesposobnosti supresije ACTH, s posljedičnom hiperandrogenemijom koja izaziva virilizaciju i smanjenu brzinu rasta u djece te u konačnici do prijevremenog prestanka rasta. Nadzor uključuje mjerenje razine 17–hidroksiprogesterona i DHEA ili androstenediona u serumu, kao i određivanje brzine rasta i sazrijevanja kostiju svake godine.
Za oblik s gubitkom soli, uz kortikosteroide, liječenje održavanja uključuje nadomjestak mineralokortikoida za obnovu homeostaze natrija i kalija. Ako dođe do gubitka soli, oralno se primjenjuje fludrokortizon (obično 0,1 mg jednom dnevno, u rasponu od 0,05 do 0,3 mg). Dojenčadi je obično potrebno oralno nadomještanje soli otprilike godinu dana. Pažljiv nadzor tijekom liječenja je od ključne važnosti.
Kod akutne bolesti, kortikosteroidne doze se povećavaju (obično dvostruko ili trostruko) kako bi se spriječila adrenalna kriza. Mineralkortikoidna supstitucija se ne podešava. Kada je oralna primjena nepouzdana (npr. povraćanje ili životno ugrožavajuća situacija), može se primijeniti IM injekcija hidrokortizon (50 do 100 mg/m2). Kada se terapija primjenjuje parijenteralno, djeca obično trebaju nadzor u hitnoj službi kako bi se utvrdilo je li im je potrebna IV tekućina, dodatni kortikosteroidi, ili oboje.
Ženskoj djeci može biti potrebna kirurška rekonstrukcija s redukcijskom klitoroplastikom i konstrukcijom otvora rodnice. Često je u odrasloj dobi potrebna dodatna operacija. Uz odgovarajuću njegu i pozornost na psihoseksualne probleme, može se očekivati normalna spolna funkcija i plodnost.
Za prenatalno liječenje, kortikosteroid (obično deksametazon) daje se majci kako bi se suzbila fetalna hipofizna sekrecija ACTH i tako smanjila ili spriječila maskulinizacija pogođenih ženskih fetusa. Liječenje, koje je pokusno, mora započeti u prvih nekoliko mjeseci gestacije.
Liječenje neklasičnog 21-hydroxylase deficiency ovisi o simptomima. Liječenje nije potrebno ako bolest asimptomatska. Ako je simptomatska, liječenje kortikosteroidima je slično kao i kod klasičnog manjka 21-hidroksilaze, a niže doze su često učinkovite. Mineralkortikoidna supstitucija nije potrebna.
Tjelesna površina (Du Boisova metoda)
Ključne točke
-
Djeca s nedostatkom 21-hidroksilaze imaju različite stupnjeve suviška androgena i oko 70% ima oblik s gubitkom soli koju uzrokuje nedostatak aldosterona.
-
U žena se višak androgena obično očituje pri rođenju dvosmislenim vanjskim genitalijama (npr. povećanje klitorisa, spajanje velikih usana, urogenitalni sinus umjesto jasnih uretralnih i vaginalnih otvora); kasnije u životu mogu imati hirzutizam, oligomenoreju i akne.
-
Kod muškaraca, višak androgena možda se neće uočiti ili se može očitovati u djetinjstvu s povećanom brzinom rasta i ranim znakovima puberteta.
-
U oba spola, oblik s gubitkom soli uzrokuje hiponatrijemiju i hiperkalemiju.
-
Dijagnoza se postavlja određivanjem steroidnih hormona, a ponekad i stimulacijom ACTH i/ili genotipizacijom.
-
Liječiti supstitucijom kortikosteroida, a ponekad i mineralokortikoida; ženski spol može zahtijevati rekonstruktivnu kirurgiju.