IMUNOLOŠKE, ALERGOLOŠKE I REUMATSKE BOLESTI
Osnovni dijagnostički postupci u reumatologiji, imunologiji i alergologiji
Primarne imunodeficijencije
Reakcije preosjetljivosti, alergije i autoimunost
Alergijski rinokonjuktivitis, urtikarija i angioedem
Anafilaksija i anafilaktički šok
Hereditarni angioedem
Mastocitoze
Autoupalne bolesti
Hemofagocitni sindrom
Reumatoidni artritis
Sistemski eritemski lupus
Antifosfolipidni sindrom
Sistemska skleroza
Sjögrenov sindrom
Polimiozitis i dermatomiozitis
Miješana bolest vezivnoga tkiva
Sindrom vaskulitisa
Seronegativni spondiloartritisi
Relapsirajući polihondritis
IgG4 bolest
Osteoartritis
Infektivni artritis i koštane infekcije
Artropatije uzrokovane odlaganjem kristala
Fibromialgija
Kompleksni regionalni bolni sindrom
Amiloidoza
Literatura
OSNOVNI DIJAGNOSTIČKI POSTUPCI U REUMATOLOGIJI, ALERGOLOGIJI I IMUNOLOGIJI
Osnovu dijagnostike u reumatologiji, imunologiji i alergologiji čine laboratorijski nalazi (opći, specifični), specifične alergološke metode te slikovne dijagnostičke metode.
OPĆI LABORATORIJSKI NALAZI
Kao i kod drugih bolesti, pri inicijalnoj se evaluaciji koristimo standardnim laboratorijskim nalazima koji uključuju kompletnu i diferencijalnu krvnu sliku, koagulogram te osnovne biokemijske pretrage (elektroliti, pokazatelji bubrežne i jetrene funkcije, mišićni enzimi i dr.). U dijagnostici ove skupine bolesti važniju ulogu mogu imati vrijednosti sedimentacije eritrocita i C-reaktivnoga proteina, broj leukocita te eritrocita i hemoglobina. Povišene vrijednosti C-reaktivnoga proteina i ubrzana sedimentacija eritrocita ukazuju na upalnu komponentnu bolesti i odraz su aktivnosti bolesti. U tumačenju broja leukocita najviše se vodimo diferencijalnom krvnom slikom: neutrofilija je prisutna kod bakterijskih infekcija (septički artritis, osteomijelitis), međutim, dio je i kliničke slike autoupalnih sindroma, limfopenija se susreće kod bolesnika s virusnim infekcijama, a može biti prisutna i kod bolesnika sa sistemskim eritemskim lupusom, dok eozinofiliju možemo susresti uslijed alergijskih bolesti. Trombocitozu se može susresti kod bolesnika s kroničnom infekcijom (tzv. reaktivna, sekundarna trombocitemija), dok je trombocitopenija znak aktivnosti nekih bolesti (kao što su sistemski eritemski lupus i antifosfolipidni sindrom), ali može biti i posljedica mijelosupresije izazvane primjenom lijekova. Kod većine bolesnika s kroničnim upalnim (autoimunim) bolestima pojavljuje se anemija kronične bolesti (normocitna, normokromna) te deplecija željeza. Kod manjega broja može se naći i sideropenična anemija kao rezultat okultnoga krvarenja iz probavnoga sustava, moguće uslijed kronične primjene nesteroidnih protuupalnih lijekova. Patološke promjene na kostima rezultiraju porastom vrijednosti alkalne fosfataze. Određivanje vrijednosti urata indicirano je kod bolesnika sa sumnjom na urički artitis (giht). Elektroforeza proteina seruma i/ili urina indicirana je u diferencijalno dijagnostičke svrhe (isključenje multiploga mijeloma), ali je i prvi korak u detektiranju hipogamaglobulinemije.
SPECIFIČNI LABORATORIJSKI NALAZI
Specifične laboratorijske pretrage u reumatologiji, imunologiji i alergologiji obuhvaćaju određivanje serumskih autoantitijela (reumatoidni faktor, anticitrulinska antitijela, antinuklearna antitijela, antitijela na dvostruku uzvojnicu DNA, antitijela na ekstraktibilne nuklearne antigene, antineutrofilna citoplazmatska antitijela, antifosfolipidna antitijela), te specifičnih komponenata imunološkoga sustava (ukupni i specifični imunoglobulini, komponente komplementa).
Reumatoidni faktor (RF) predstavlja autoantitijelo (obično IgM, rjeđe IgG ili IgA) usmjereno na Fc fragment imunoglobulina G (IgG). On se otkrije kod više od 70 % bolesnika koji boluju od reumatoidnoga artritisa (RA), ali ona nisu dijagnostička i specifična za RA, s obzirom na to da se mogu naći i u drugim autoimunim reumatološkim bolestima (npr. sistemski eritemski lupus, Sjögrenov sindrom, sistemska skleroza), kroničnim infekcijama (tuberkuloza, infektivni endokarditis, hepatitis) te kod starijih osoba bez posebnih simptoma. Normalne vrijednosti RF-a su manje od 14 IU/mL.
Anticitrulinska antitijela (anti-CCP) predstavljaju autoantitijela usmjerena na ciklički citrulinizirani peptid, a mogu se naći kod više od 80 % osoba koje boluju od reumatoidnoga artritisa za što imaju visoku specifičnost (90 %). Posebnu korist pokazuju kod bolesnika koji imaju negativan reumatoidni faktor, sa svrhom razlikovanja reumatoidnoga artritisa od akutnoga prolaznog sinovitisa. Pozitivan RF i anti-CCP povezani su lošijom prognozom i većom predikcijom nastanka koštanih erozija kod bolesnika s reumatoidnim artritisom. Normalne vrijednosti anti-CCP-a su manje od 5,0 IU/mL.
Antinuklearna antitijela (ANA) su heterogena skupina autoantitijela usmjerenih na funkcijske i strukturne dijelove stanične jezgre. Najčešće se koriste u testu probira na sistemski eritemski lupus, sistemsku sklerozu i miješanu bolest vezivnoga tkiva (engl. mixed connective tissue disease, MCTD) jer negativna ANA antitijela čine navedene dijagnoze malo vjerojatnima. Ona se mogu se naći i uslijed drugih autoimunih bolesti, kao što su Sjögrenov sindrom, polimiozitis i dermatomiozitis te autoimuni hepatitis, ali i kod infekcija, upala, neoplazmi, pa čak i kod zdravih pojedinaca, stoga je preporuka pretragu raditi samo kada postoji sumnja na upalnu reumatsku bolest. Nalaz ANA-e se izražava u titru i tipu florescencije. Najčešći tipovi fluorescencija su točkasta, homogena ili nuklearna koje nalazimo kod sistemskoga eritemskog lupusa, te anticentromerna ili zrnata nuklearna kod sistemske skleroze. Normalne vrijednosti ANA titra su manje od 1:80.
Antitijela na dvostruku uzvojnicu DNA (anti-dsDNA) predstavljaju podvrstu antinuklearnih antitijela koja su usmjerena na dvolančanu DNA molekulu. Ona su visoko specifična za sistemski eritemski lupus. S obzirom na to da njihove vrijednosti koreliraju s aktivnošću bolesti, ta se antitijela mogu koristiti i u praćenju odgovora na liječenje, a ne samo u dijagnostici. Povišene vrijednosti mogu se naći i kod kroničnoga hepatitisa, primarne bilijarne ciroze te infektivne mononukleoze. Dobiveni rezultat interpretira se kao negativan (vrijednosti manje od 4 IU/mL), slabo pozitivan (vrijednosti 4 do 6 IU/mL) ili pozitivan (vrijednosti veće od 10 IU/mL).
Antitijela na ekstraktibilne nuklearne antigene (ENA) označavaju skupinu specifičnih antitijela usmjerenih na određene jezgrene antigene koji su povezani s određenim autoimunim bolestima (tzv. ENA screen): anti-SS-A ili anti-Ro (Sjögrenov sindrom, sistemski eritemski lupus), anti-SS-B ili anti-La (Sjögrenov sindrom, sistemski eritemski lupus), anti-RNP/Sm (miješana bolest vezivnoga tkiva, sistemski eritemski lupus, Sjögrenov sindrom, progresivna sistemska skleroza), anti-Jo-1 (polimiozitis, dermatomiozitis), anti-Scl-70 (progresivna sistemska skleroza) te anti-U1-nRNP (nediferencirana bolest vezivnoga tkiva). U ENA probiru rezultati se označavaju kao pozitivni ili negativni.
Antineutrofilna citoplazmatska antitijela (ANCA) predstavljaju autoantitijela klase IgG usmjerena protiv granula lizosoma neutrofila i makrofaga, a razlikujemo dvije podvrste: autoantitijela usmjerena na proteinazu 3 koja se nazivaju citoplazmatska ANCA antitijela (cANCA ili PR3) i autoantitijela usmjerena na mijeloperoksidazu koja se nazivaju perinuklearna ANCA antitijela (pANCA ili MPO). cANCA i pANCA povezani su s vaskulitisima malih krvnih žila: mikroskopski poliangitis (MPA), eozinofilna granulomatoza s poliangitisom (EGPA, stari naziv Churg-Straussov sindrom), granulomatoza s poliangitisom (GPA, stari naziv Wegenerova granulomatoza).
Antifosfolipidna antitijela su skupina autoantitijela koja imaju sposobnost vezanja za fosfolipidne ostatke na membranama endotelnih stanica, trombocita, stranica trofoblasta ili fosfolipida vezanih za faktore koagulacije čime se mijenja njihov fiziološki koagulacijski učinak i stvara prokoagulantno stanje. U kliničkoj se praksi određuju lupus antikoagulans (LAC), antikardilipinska antitijela (ACL) te anti-β2-glikoprotein-I antitijela (aβ2GP-I). Pozitivna antifosfolipidna antitijela nalazimo uslijed antifosfolipidnoga sindroma.
Komponentne komplementa C3 i C4 predstavljaju standardni dio laboratorijske obrade bolesnika sa sumnjom na autoimunu bolest vezivnoga tkiva. Povišene vrijednosti C3 i C4 nalazimo pri akutnim upalnim zbivanjima te kroničnim malignim i nemalignim bolestima (amiloidoza, šećerna bolest), dok snižene vrijednosti mogu biti posljedica urođenoga nedostatka C3 i/ili C4 ili utroška uslijed prisutnosti cirkulirajućih imunokompleksa koji ih aktiviraju i troše (npr. aktivna faza sistemskoga eritemskog lupusa).
Ukupni i specifični imunoglobulini. IgA, IgG i IgM najčešće se skupa određuju, njihove su vrijednosti povišene pri infekcijama, autoimunim bolestima te bolestima jetre, a kada su snižene znak su primarne ili sekundarne imunodeficijencije. Određivanje ukupnoga IgE-a korisno je u dijagnostici alergoloških oboljenja (povišen u stanjima atopije). Moguće je određivati i vrijednosti specifičnoga IgE antitijela za pojedine alergene RAST-metodom (radio-alergo-sorbent test).
ALERGOLOŠKA DIJAGNOSTIKA
In vitro alergološki testovi podrazumijevaju laboratorijsko određivanje ukupne vrijednosti imunoglobulina E te vrijednosti specifičnih imunoglobulina E (IgE), mjerenje upalnih stanica (eozinofili u perifernoj krvi i brisu nosa, mjerenje specifičnih T-limfocita) te mjerenje citokina i medijatora oslobođenih u alergijskim reakcijama (eozinofilni kationski protein, mastocitna triptaza, histamin). Povišene vrijednosti ukupnoga IgE-a, eozinofilija te povišene vrijednosti medijatora alergijske reakcije ukazuju na alergijsko zbivanje, dok određivanje specifičnoga IgE-a i specifičnih T-limfocita omogućuju otkrivanje alergena koji dovodi do alergijske reakcije.
In vivo alergološki testovi podrazumijevaju primjenu kožnih testova kojima se testira osjetljivost osoba na pojedine nutritivne, inhalacijske ili neke druge antigene. U tu se svrhu najčešće primjenjuju epikutani test (engl. patch), ubodni (engl. prick) test te test ogrebotine (engl. scratch). Testovi podrazumijevaju nanošenje alergena na kožu putem posebnih naljepaka ili u kožu bockanjem ili zagrebavanjem površine kože na koju je nanešena otopina alergena te očitavanjem kožne reakcije na njih. Najčešće izvođeni provokacijski test je bronhoprovokacijski test koji je opisan u poglavlju o „Dijagnostički postupci u pulmologiji“.
SLIKOVNE I OSTALE DIJAGNOSTIČKE METODE
Radiološke dijagnostičke metode. Klasično radiografsko snimanje (nativno) najčešće predstavlja osnovnu i ishodišnu slikovnu dijagnostičku metodu u obradi bolesnika s poremećajem koštano-zglobnoga sustava. Ono omogućava uvid u strukturalne promjene koštanih struktura, ali se njime ne dobivaju značajniji podaci o mekotkivnim strukturama, kao što su hrskavica i zglobne sveze. U ranoj fazi bolesti radiološki se može otkriti samo zadebljanje zglobne čahure, što nema veće diferencijalno-dijagnostičko značenje. Prve specifičnije promjene javljaju se nekoliko mjeseci od početka bolesti. Detaljnu morfologiju navedenih mekotkivnih struktura omogućava magnetna rezonanca (MRI), a predstavlja i znatno osjetljiviju metodu u ranim fazama bolesti koštano-zglobnoga sustava kada klasične radiografske snimke pokazuju uredan nalaz. U dijagnostici promjena mekotkivnih zglobnih struktura sve više se koristi i ultrazvučni pregled (edem zglobnih sveza, izljev, sinovitis). Scintigrafija koštanoga sustava i zglobova korisna je za prikazivanje upalnih zbivanja i patoloških procesa praćenih pojačanom koštanom pregradnjom (npr. metastaze). Koštana denzitometrija (DXA) koristi se za procjenu gustoće kostiju što najveću ulogu ima u dijagnostici osteoporoze.
Punkcija i analiza sinovijalne tekućine (artrocenteza) najčešće se koristi u dijagnostici septičkoga i reaktivnoga artritisa te artritisa izazvanoga nakupljanjem kristala. Sama pojava povećane količine tekućine u zglobu znak je patološkoga zbivanja, a njezina biokemijska, mikrobiološka i citološka analiza ukazuju na narav toga patološkog zbivanja. Normalna sinovijalna tekućina je bistra i bezbojna te sadrži manje od 3000 leukocita u mm3. U upalnim stanjima broj leukocita je veći od 3000/mm3, a u septičkom artritisu raste i preko 50 000/mm3. Osim u dijagnostičke svrhe, ona može poslužiti i kao terapijska metoda (aspiracija tekućine dovodi do smanjenja intraartikularnoga tlaka i osjeta bolnosti, te za uklanjanje viška kristala).
Citološke i histološke metode također su važne u reumatološkoj dijagnostici. Citološki se najčešće analizira uzorak sinovijalne tekućine dobivene punkcijom zgloba što je prethodno spomenuto. Histološke pretrage imaju značajnu ulogu u dijagnostici autoimunih bolesti i vaskulitisa kada se uzima uzorak zahvaćenoga tkiva ili organa (najčešće koža ili bubrezi) te analizira metodama svjetlosne, elektronske i imunofluorescenscijske mikroskopije.
PRIMARNE IMUNODEFICIJENCIJE
Uvod. Imunodeficijencija označava stanje koje se odnosi na nedostatak ili poremećaj funkcije imunološkoga sustava što rezultira povećanom sklonošću prema razvoju infekcija i malignih oboljenja, a prema porijeklu se dijeli na primarne i sekundarne. Primarne imunodeficijencije nastaju uslijed urođenoga (nasljednog) poremećaja imunološkoga sustava, dok su sekundarne imunodeficijencije stečene i nastaju kao posljedica različitih bolesti i stanja koja narušavaju funkciju imunološkoga sustava, kao što su infekcije, maligna oboljenja i druge kronične nezarazne bolesti, kemoterapijsko liječenje ili imunosupresivno liječenje. Primarne imunodeficijencije predstavljaju rijetka oboljenja s procijenjenom učestalošću od oko 1:4000 osoba unutar opće populacije, a kako se radi o nasljednim oboljenjima, u većini se slučajeva dijagnoza postavlja već tijekom djetinjstva.
Klasifikacija. Primarne imunodeficijencije možemo podijeliti u nekoliko skupina ovisno o temeljnom poremećaju: poremećaji funkcije T-limfocita, poremećaji funkcije B-limfocita, kombinirani poremećaji T- i B-limfocita, poremećaji funkcije neutrofila te poremećaji funkcije komplementa. Najčešći oblici primarnih imunodeficijencija koje susrećemo u odrasloj dobi su selektivna deficijencija imunoglobulina A te obična varijabilna imunodeficijencija, pa su oni posebno izdvojeni i opisani. Većina ostalih imunodeficijencija očituje se rano u djetinjstvu ili dojenačkoj dobi te su predmet izučavanja pedijatrije.
SELEKTIVNA DEFICIJENCIJA IMUNOGLOBULINA A
Selektivna deficijencija imunoglobulina A (IgA) predstavlja najčešći oblik primarne imunodeficijencije čija se prevalencija procjenjuje na 1:500 do 1:700 unutar opće populacije, s time da je većina oboljelih asimptomatska i zato ostaje neprepoznata i nedijagnosticirana. Radi se o nasljedno uvjetovanome poremećaju čiji genski defekt i molekularna podloga bolesti još uvijek nisu poznati (nedostatak faktora koji utječu na sintezu i ekspresiju IgA unutar B-limfocita). Postoje i oblici bolesti za koje nije dokazana obiteljska pojavnost, a često se povezuju s primjenom antiepileptika, posebice fenitoina. U kliničkoj slici najčešće nalazimo recidivirajuće i kronične infekcije dišnoga sustava (sinusitis, otitis, traheobronhitis) koje su karakteristično uzrokovane enkapsuliranim bakterijama, kao što su H. influenzae i S. pneumoniae. Oboljeli pokazuju i povećanu sklonost prema razvoju atopijskih i autoimunih oboljenja (Gravesova bolest, sistemski eritemski lupus, juvenilni reumatoidni artritis, šećerna bolest tip I, celijakija, Adidisonova bolest, itd.), ali i razvoju malignih bolesti (adenokarcinom želuca i crijeva, Hodgkinova bolest, akutna limfoblastična leukemija, itd). Dijagnoza se postavlja na osnovi nalaza sniženih vrijednosti IgA-a (IgA < 7mg/dL) uz uredne vrijednosti IgG-a i IgM-a te urednu vrijednost i funkciju T-limfocita. Kod manjega broja oboljelih mogu se naći i specifična anti-IgA antitijela, što može biti povezano s povećanim rizikom od razvoja anafilaksije nakon primjene krvnih derivata ili humanih imunoglobulina. Specifično liječenje imunoglobulinima nije potrebno, kod većine bolesnika kontrola bolesti postiže se odgovarajućim mjerama zaštite od infekcije te ranom primjenom antibiotika u slučaju razvoja infekcije.
OBIČNA VARIJABILNA IMUNODEFICIJENCIJA
Obična varijabilna imunodeficijencija (engl. common variable immunodeficiency - CVID) predstavlja heterogenu skupinu poremećaja koje karakterizira povećana učestalost infekcija, autoimunih te malignih bolesti, a obilježena je deficijentnom produkcijom više od jedne različite velike klase protutijela. B-stanice kod CVID-bolesnika mogu prepoznati antigen i odgovoriti proliferacijom, ali postanu kvantitativno oštećene u sposobnosti da postanu memorijske B-stanice i zrele plazma stanice. Procijenjena prevalencija ovoga poremećaja iznosi od 1:25 000 do 1:50 000 unutar opće populacije s podjednakom pojavnošću kod oba spola. Iako se bolest može manifestirati u bilo kojoj životnoj dobi, najčešće se to događa u ranoj odrasloj dobi (drugo i treće desetljeće života). Kod većine bolesnika bolest se javlja sporadično, a obiteljsko pojavljivanje bilježi se kod oko 10 do 20 % oboljelih. Glavne kliničke manifestacije predstavljaju sklonost bakterijskim infekcijama dišnoga i probavnog sustava (otitis, sinusitis, bronhitis, pneumonije, gastroenteritis, malapsorpcija) uz povećanu učestalost razvoja autoimunih i malignih bolesti. Glavni kriteriji za postavljanje dijagnoze ovoga poremećaja su: 1) snižena vrijednost IgG-a uz koju često nalazimo i snižene vrijednosti IgM-a i IgA-a, 2) neodgovarajući odgovor na primjenu cjepiva (izostanak stvaranja specifičnih antitijela nakon provedenoga cijepljenja te 3) isključeni drugi mogući uzroci hipogamaglobulinemije. Broj B-limfocita kod tih je bolesnika većinom uredan. Liječenje se sastoji od rane primjene antibiotika širokoga spektra pri pojavi kliničke sumnje na infekciju kao i prevencije supkutanom ili intravenskom primjenom humanih imunoglobulina u mjesečnoj dozi od 300 do 600 mg/kg.
OSTALI OBLICI PRIMARNIH IMUNODEFICIJENCIJA
DiGeorgeov sindrom rijetka je kongenitalna anomalija karakterizirana u najvećem broju slučajeva mikrodelecijom 22. kromosoma koja uzrokuje poremećaj razvoja trećega i četvrtoga ždrijelnog (škržnog) luka tijekom fetalnoga razvoja. Bolest se naslijeđuje autosomno-dominantno, međutim, najčešće se radi o de novo mutaciji uslijed utjecaja vanjskih teratogenih čimbenika. Budući da se iz te embrionalne osnove razvijaju paratireoidne žlijezde, luk aorte i timus, posljedično dolazi do njihove ageneze ili hipogeneze što rezultira hipoparatireoidizmom (hipokalcemična tetanija), poremećajima razvoja srca i aorte (anomalije) te deplecijom T-limfocita s obzirom na nedostatak timusa, mjesta na kojemu dolazi do njihova potpunog sazrijevanja. Kod ove djece često je prisutna i dismorfija lica te rascjepi nepca. Recidivirajuće (oportunističke) infekcije javljaju se ubrzo nakon rođenja. Dijagnoza se postavlja na osnovi kliničke slike (učestale infekcije, tetanija, dismorfija lica), laboratorijskih nalaza (hipokalcijemija, smanjen broj T-limfocita) te slikovnih metoda kojima potvrđujemo nedostatak timusa i anomalije srca i aorte. Osnovu liječenja čini suplementacija kalcija, kirurška korekcija postojećih srčanih i aortalnih anomalija te profilaktička primjena antibiotika. U novije vrijeme provodi se i transplantacija timusa korištenjem tkiva fetusa ili matičnih stanica od HLA-identičnih braće ili sestara.
X-vezana agamaglobulinemija ili Brutonova agamaglobulinemija predstavlja oblik poremećaja stvaranja i funkcije B-limfocita koja nastaje uslijed mutacija na X-kromosomu gdje nalazimo gen za tirozin kinazu koja je uključena u proces stanične aktivacije i sazrijevanje B-limfocita. S obzirom na to da se radi o poremećaju X-kromosoma, od ovoga poremećaja obolijevaju samo osobe muškoga spola. Kliničke manifestacije u vidu recidivirajućih bakterijskih i virusnih infekcija javljaju se obično nakon 6. mjeseca života (do tada su djeca zaštićena majčinim protutijelima). U laboratorijskim nalazima nalazimo sniženje imunoglobulina uz odsutnost B-limfocita, dok su broj i funkcija T-limfocita uredni. Biopsija limfnih čvorova ima karakterističan nalaz koji uključuje hipoplaziju te nedostatak limfnih folikula i germinativnih centara. Osnovu liječenja čini supstitucija imunoglobulina uz učestale primjene antibiotika. Usprkos tim mjerama, većina oboljelih razvija kronične infekcije, a postoji i veći rizik od razvoja malignih bolesti.
Teška kombinirana imunodeficijencija predstavlja rijedak oblik primarne imunodeficijencije koju karakterizira odsutnost T-limfocita uz normalan ili smanjen broj B-limfocita i NK-stanica. Opisano je više genskih defekata i potencijalnih molekularnih mehanizama u razvoju ovoga poremećaja kao što su nedostatak γ-lanca receptora interleukina 2, manjak adenozin-deaminaze te nedostatak α-lanca receptora interleukina 7. Kliničke manifestacije ovoga poremećaja javljaju se već tijekom dojenačkoga perioda u vidu učestalih gljivičnih i bakterijskih infekcija, a ovisno o genskom defektu mogu se naći i druge manifestacije kao što su eksfolijativni dermatitis (Omennov sindrom) ili abnormalnosti kostiju kod nedostatka adenozin-deaminaze. Dijagnoza se postavlja na osnovi kliničke slike i laboratorijskih nalaza, a ako se ne započne promptno liječenje intravenskim imunoglobuinima, smrtnost od ovoga poremećaja je visoka. Jedini dostupni terapijski postupak koji može dovesti do izliječenja je transplantacija matičnih stanica od HLA-identičnoga donora. Kod dokazane deficijencije adenozin-deaminaze primjenjuje se supstitucijsko liječenje, a sve više uspjeha u kliničkim istraživanjima postiže i liječenje genskom terapijom.
Hiper-IgM sindrom predstavlja oblik primarne imunodeficijencije kojega obilježava pojačana produkcija imunoglobulina M uz istodobni nedostatak ostalih oblika imunoglobulina. Radi se o poremećaju koji najčešće nastaje uslijed mutacije gena za CD154 ili CD40 ligand koji se nalazi na površini aktiviranih pomoćnićkih T-stanica, a djeluje na B-limfocite tako što skreće proizvodnju s IgM-a na proizvodnju IgA-a, IgG-a ili IgE-a. Često ovi bolesnici imaju i tešku neutropeniju, hipoplaziju limfnoga tkiva te povećanu sklonost razvoju limfoma i autoimunih oboljenja. Dijagnoza se postavlja na temelju kliničke slike i laboratorijskih nalaza, a liječenje se sastoji od redovne primjene intravenskih imunoglobulina. Autoimune i limfoproliferativne komplikacije mogu dobro odgovoriti na anti-CD20 terapiju (rituksimab). Bolest u načelu ima lošu prognozu i većina bolesnika umre tijekom djetinjstva od posljedica bakterijskih ili drugih komplikacija.
Poremećaji funkcije fagocita. Najznačajniji poremećaji iz ove skupine su kronična granulomska bolest i adhezijski poremećaji leukocita (bolesti lijenih leukocita). Kronična granulomatozna bolest obilježena je smanjenom baktericidnom sposobnosti leukocita uslijed smanjenoga stvaranja slobodnih kisikovih radikala (vodikov peroksid, superoksid), zbog čega su bolesnici skloni razvoju bakterijskih infekcija što se očituje već tijekom djetinjstva. Bolest počinje stvaranjem apscesa koji postupno poprimaju značajke kronične infekcije i postaju granulomi, a zahvaćeni mogu biti praktično svi organski sustavi. Dijagnoza se postavlja na temelju specifičnih funkcionalnih testova procjene funkcije fagocita (sposobnost oksidativnoga metabolizma - "respiratorni prasak"). Liječenje se sastoji od kontinuirane antimikrobne profilakse uz povremenu primjenu interferona u svrhu imunomodulacije, a opcija je i liječenje transplantacijom matičnih stanica od HLA-podudarajućega donora, a uspjeh pokazuje i nova genska terapija. Adhezijski poremećaji leukocita nastaju kao posljedica poremećaja gena koji kodiraju adhezivne glikoproteine na površini leukocita što omogućava njihovu adheziju na mjestu upale, odnosno dijapedezu i mogućnost migracije u ekstravaskularni prostor zbog čega nastaju teške gnojne infekcije. Dijagnoza se postavlja dokazivanjem manjka adhezivnih glikoproteina na površini leukocita uporabom specifičnih protutijela i postupkom protočne citometrije. Liječenje podrazumijeva kontinuiranu antimikrobnu profilaksu. Prognoza oba poremećaja je teška i većina oboljele djece umre u djetinjstvu od posljedica infektivnih komplikacija.
Poremećaji funkcije sustava komplementa rijetki su u kliničkoj praksi. Opisane su deficijencije gotovo svih komponenata komplementa, a najčešće su deficijencije komponenata C2, C1q i C3 koje su povezane s hipogamaglobulinemijom i nastankom teških infekcija. Bolesnici imaju veću incidenciju autoimunih bolesti, osobito sistemskoga eritemskog lupusa. U ovu skupinu bolesti ubraja se i hereditarni angioedem koji nastaje uslijed nedostatka ili smanjene aktivnosti inhibitora C1, koji je posebno opisan.
REAKCIJE PREOSJETLJIVOSTI, ALERGIJE I AUTOIMUNOST
Uvod. Imunološka preosjetljivost predstavlja prekomjernu i neprimjerenu imunološku reakciju na antigen koja može izazvati štetan upalni odgovor i oštećenje tkiva. Ona se može razviti kao odgovor na neki vanjski antigen, kada govorimo o alergijama, ili na vlastiti antigen, kada govorimo o autoimunim reakcijama. U načelu razlikujemo četiri oblika imunološke preosjetljivosti: 1) alergijsko-anafilaktička preosjetljivost (tip I), citotoksična preosjetljivost posredovana protutijelima (tip II), preosjetljivost posredovana imunokompleksima (tip III) te stanično posredovana preosjetljivost (tip IV).
Alergijsko-anafilaktička preosjetljivost (reakcija preosjetljivosti tipa I) javlja se kod osoba koje su prethodno bile u dodiru s antigenom na kojega su razvile preosjetljivost, tj. senzibilizirane su na njega te se on naziva alergenom. Kod prvoga kontakta ključnu ulogu imaju AP-stanice (predočne stanice, APC, engl. antigen presenting cell) koje navedeni antigen prezentiraju limfocitima CD4+ koji putem citokina senzibiliziraju B-limfocite na stvaranje specifičnih IgE antitijela koja se vežu na površinu bazofila i mastocita te tako cirkuliraju u krvi. Prilikom sljedećega kontakta navedeni antigen (alergen) vezat će se na stvorena IgE antitijela koja se nalaze na površini mastocita i bazofila, a ta interreakcija djelovat će kao signal za pokretanje unutarstaničnih signala koji će dovesti do njihove degranulacije prilikom čega se iz njih otpuštaju različiti vazoaktivni posrednici. To vezanje odvija se po principu da se jedna molekula antigena mora vezati za dvije molekule IgE te ih tako premostiti. Ta se reakcija odvija u dvije faze: rana i kasna. Rana faza započinje unutar 5 do 30 minuta od kontakta s antigenom (alergenom) i karakterizirana je otpuštanjem već stvorenih posrednika koji se nalaze u sekrecijskim zrncima mastocita i bazofila (histamin, faktori kemotaksije, neutralne proteaze, heparin), a kasna faza nastupa kroz 2 do 24 sata, a izazivaju je tzv. sekundarni posrednici koji se počinju stvarati tek nakon podražaja, a radi se o derivatima arahidonske kiseline (leukotirijeni, prostaglandini) te različiti citokini i kemokini. Navedeni posrednici djeluju tako da povećavaju propusnost krvnih žila, dovode do kontrakcije glatko-mišićne muskulature (osobito dišnih puteva), povećavaju sekrecijsko djelovanje mukoznih žlijezda, aktiviraju sustav komplementa, privlače druge imunostanice na mjesto događaja i sl. (Tablica 9.1.). Reakcije preosjetljivosti tipa I mogu biti lokalizirane i sistemske, a najčešći klinički primjeri su alergijski rinokonjuktivitis, alergijska astma, urtikarija i anagioedem te anaflaksija i anafilaktički šok.
|
Tablica 9.1. Medijatori reakcije preosjetljivosti tipa I
|
|
Medijatori rane faze
|
|
Histamin i serotonin (bronhokonstrikcija, povećana vaskularna propusnost)
Faktori kemotaksije neutrofila i eozinofila (infiltracija upalnim stanicama)
|
|
Medijatori kasne faze
|
|
Leukuotrijen B4 (kemotaksija upalnih stanica)
Leukotrijeni C4, D4 i E4 (bronhokonstrikcija, edem)
Prostaglandini, trombkosan (produljena hiperreaktivnost dišnih puteva)
|
Citotoksična preosjetljivost posredovana protutijelima (reakcija preosjetljivosti tipa II) nastaje kao posljedica interreakcije između IgG ili IgM protutijela s endogenim ili egzogenim antigenima koji se nalaze na površini stanica, membrana ili u ekstracelarnom prostoru. Oštećenje tkiva ovom reakcijom može nastati na jedan od tri načina: 1) vezanje IgG ili IgM protutijela za navedene antigene može dovesti do aktivacije sustava komplementa posredovano Fc receptorom što rezultira direktnom citolizom ili opsonizacijom stanice pri čemu ona postaje podložna fagocitozi; 2) vezanjem IgG ili IgM antitijela za navedene antigene također može dovesti do direktne fagocitoze posredovane stanicama koje imaju Fc receptor (monociti, neutrofili, NK-stanice) te 3) vezane IgG ili IgM protutijela za navedene antigene može aktivirati različite unutrastanične signale koji će dovesti do poremećaja u funkciji ili oštećenja stanice i bez nastanka upalne reakcije. Primjeri bolesti koje nastaju ovim putem su transfuzijska reakcija, autoimune hemolitičke anemije, neki oblici glomerulonefritisa, Gravesova bolest, mijastenija gravis i dr.
Preosjetljivost posredovana imunokompleksima (reakcija preosjetljivosti tipa III) nastaje uslijed stvaranja imunokopleksa koji se odlažu u različita tkiva i organe gdje dovode do aktivacije sustava komplementa koji izaziva njihovo oštećenje. Imunokompleksi predstavljaju komplekse koje čine antigen (endogeni ili egzogeni) i specifično antitijelo s kojim se spaja, a mogu nastati u cirkulaciji i odlagati se u različite organe i tkiva (sistemska reakcija) ili se mogu stvarati na mjestu odlaganja antigena (lokalizirana reakcija). Neki od primjera bolesti koje nastaju ovim putem su serumska bolest, sistemski eritemski lupus, određeni oblici glomerulonefritisa i dr.
Stanično posredovana preosjetljivost (reakcija preosjetljivosti tipa IV) posredovana je imunološkim stanicama, u prvom redu T-limfocitima. Ona podrazumijeva dva oblika reakcija: reakcija odgođene preosjetljivosti u kojoj glavnu ulogu imaju CD4+ T-limfociti te T-stanično posredovana citotoksičnost u kojoj glavnu ulogu imaju CD8+ T-limfociti. U reakciji odgođene preosjetljivosti glavnu ulogu imaju naivni CD4+ T-limfociti koji se senzibiliziraju za određeni antigen prilikom prvoga kontakta s njima preko AP-stanica. Oni se pod utjecajem IL-12 pretvaraju u Th1 stanice koje ostaju cirkulirati u krvi i pri drugom kontaktu s istim antigenom dolazi do njihove aktivacije s posljedičnim lučenjem INF-γ, IL-2 i TNF-α koji dovode do upalne reakcije i oštećenja tkiva, a ako se antigen ne može razgraditi i dalje perzistira, dolazi do transformacije makrofaga u epiteloidne stanice pri čemu nastaju granulomi. Primjeri bolesti koje nastaju ovim putem su tuberkuloza, sarkoidoza i kontaktni dermatitis. U drugom obliku reakcije glavnu ulogu imaju CD8+ T-limfociti koji imaju sposobnost izravnoga citolitičkog djelovanja vezanjem za stanice koje na svojoj površini eksprimiraju antigen za koji su senzibilizirani. To je osnovna reakcija u borbi protiv virusnih infekcija (uništenje vlastitih stanica koje su inficirane virusom i eksprimiraju njegove antigene na svojoj površini), a javlja se i kod reakcije odbacivanja presatka.
Pojam alergija. Alergija je naziv za neprikladnu i prekomjernu reakciju imunološkoga sustava na inače bezopasne antigene iz okoliša, koji nisu povezani s ozljedom ili upalom. U osnovi se najčešće radi o reakciji koja je posredovana sintezom antigen-specifičnoga imunoglobulina E, odnosno reakciji preosjetljivosti tip I što se naziva atopijom. Alergija i atopija nisu sinonimi (iako se često tako tumače) jer pojam alergija obuhvaća sve oblike imunoreakcije na neki antigen, dok se atopija odnosi samo na one oblike imunoreakcija koje su posredovane imunoglobulinom E. Svaki antigen koji može dovesti do alergijske reakcije nakon izlaganja imunološkom sustavu naziva se alergenom. Najčešće se radi o proteinima ili polisaharidima koji mogu izazvati alergijsku reakciju samostalno (pravi alergeni) ili tek nakon što se vežu za proteinske nosače (takvi se alergeni nazivaju hapteni). Smatra se da alergijske bolesti nastaju kao rezultat poremećenih genskih odnosa i vanjskih, okolišnih čimbenika. U normalnim se okolnostima nakon rođenja imunoodgovor usmjerava u pravcu Th1 stanica (Th1 odgovor) koje su odgovorne za borbu protiv infektivnih uzročnika pri čemu dolazi do istodobnoga potiskivanja Th2 stanica (Th2 odgovor) koje mogu aktivirati eozinofile i koje potiču stvaranje IgE (proalergijski učinak). Današnji zapadnjački stil života koji podrazumijeva unaprijeđene higijenske uvijete smanjuje rani kontakt organizma s infektivnim agensima što usporava razvoj Th1 odgovora i pogoduje Th2 odgovoru čime se i tumači sve veća pojavnost alergijskih oboljenja i smanjenja imunotolerancije. Procjenjuje se da od alergijskih bolesti danas boluje oko 25 % osoba u razvijenim zemljama što ih svrstava među najčešće bolesti s kojima se susrećemo u svakodnevnoj kliničkoj praksi.
Pojam autoimunosti. Autoimunost predstavlja imunološki odgovor na vlastite antigene. Procjenjuje se da od autoimunih bolesti danas boluje oko 5 % opće populacije. Premda ima dovoljno dokaza da autoimune bolesti imaju brojne etiološke čimbenike, većina ih u osnovi nastaje uslijed tri temeljna događaja: 1) uslijed promjene vlastitoga antigena (npr. pod utjecajem lijekova ili mikroba vlastiti se antigeni mogu promijeniti i premda su te promjene malene, mogu izazvati imunološku reakciju), 2) uslijed križne reaktivnosti, pri čemu imunološki sustav zbog sličnosti vlastitoga antigena sa stranim antigenom na koji je razvio odgovor reagira i na vlastiti antigen te 3) uslijed poremećaja imunotolerancije (poremećaj odgajanja limfocita da prepoznaju vlastito) te imunoregulacije (poremećaj odnosa između Th1 i Th2 odgovora). Navedene promjene nastaju uzajamnim djelovanjem genetskih i vanjskih čimbenika. Danas je jasna povezanost između autoimunih bolesti i genetskih utjecaja, a na to ukazuje već sama činjenica da su one učestalije unutar nekih obitelji ili između jednojajčanh blizanaca. Prisutnost određenih HLA-antigena može povećati sklonost razvoju autoimunih bolesti, ali postoje i HLA-antigeni koji se smatraju protektivnima i smanjuju rizik od pojavnosti ovih bolesti. Od vanjskih čimbenika u razvoju autoimunih bolesti najviše se spominju infektivni uzročnici koji mogu inducirati autoimunost molekularnom mimikrijom, poliklonskom aktivacijom ili otpuštanjem ranije sekvestriranih (skrivenih) antigena. U autoimunim bolestima oštećenje tkiva i organa može biti posredovane reakcijom preosjetljivosti II, III i IV.
ALERGIJSKI RINOKONJUKTIVITIS, URTIKARIJA I ANGIOEDEM
Uvod. Alergijski rinokonjuktivitis, urtikarija i angioedem predstavljaju najčešće oblike alergijskih oboljenja koji se susreću u internističkoj kliničkoj praksi. Alergijska astma opisana je u poglavlju pulmologije.
ALERGIJSKI RINOKONJUKTIVITIS
Definicija i epidemiologija. Alergijski rinokonjuktivitis (peludna hunjavica) predstavlja jedno od najčešćih oblika alergijskih oboljenja koje nastaje kao posljedica reakcije preosjetljivosti na alergene koji se pojavljuju na sluznici oka i nosa. Ovaj poremećaj viđa se kod 10 do 30 % odraslih osoba unutar opće populacije, a podjednako se javlja u oba spola. Ovisno o pojavnosti i uzročnom alergenu, alergijski rinokonjuktivitis može se javiti u dva oblika: sezonski i cjelogodišnji.
Etiopatogeneza. Alergijski rinokonjuktivitis nastaje kao posljedica lokalne reakcije preosjetljivosti tipa I (alergijsko-anafilaktički tip) na pelud i druge inhalacijske alergene u području sluznice nosa i/ili očnih spojnica. Najčešći alergeni koji dovode do ove reakcije su pelud trave, korova i drveća, grinje kućne prašine, proteini životinjskih dlaka te spore pljesni i gljivica. Do razvoja simptoma dolazi nakon dospijeća peludi, spora i sličnih formacija na površinu sluznice gdje ih lokalni enzimi (npr. lizozimi) razgrađuju, prilikom čega se oslobađaju proteinske čestice molekularne težine od 10 000 do 40 000 daltona koje djeluju kao alergeni i potom se vežu za ranije stvorene molekule IgE na površini mastocita i bazofila čime započinje imunološki odgovor. Akutne promjene uključuju hiperemiju, edem i hipersekreciju, dok kroničnu formu obilježava razvoj nosne polipoze. Alergijski rinokonjuktivitis kod velikoga broja bolesnika povezan je i s razvojem alergijske astme (često joj prethodi nekoliko godina ranije).
Klinička slika. Simptomi i znakovi alergijskoga rinokonjuktivitisa su karakteristični, javljaju se epizodično i lokalnoga su karaktera. Bolesnici se žale na kihanje, imaju izraženu nosnu sekreciju i suzenje uz osjećaj svrbeža i/ili pečenja u području očiju i nosa te osjećaj začepljenosti nosa. Kod nekih bolesnika izraženiji su nosni, a kod nekih očni simptomi. Često je prisutan i osjećaj svrbeža u području prednje strane vrata, a može se javiti i kašalj. Uporna nosna kongestija može ometati drenažu sinusa te dovesti do glavobolje i sinusitisa, kao i pojave upale srednjega uha (začepljenost Eustahijeve tube).
Dijagnoza se kod većine bolesnika postavlja na osnovi kliničke slike i epidemioloških podataka. Često se dobije podatak o prisutnosti istoga ili drugih atopijskih oboljenja kod bližih srodnika. U potvrdi dijagnoze od koristi su pretrage krvi (nalaz eozinofilije u diferencijalnoj krvnoj slici, povišene vrijednosti ukupnoga IgE-a) te bris nosa na eozinofile. Za identifikaciju uzročnoga alergena koristi se intradermalno alergološko testiranje (prick-test) ili određivanje specifičnih IgE protutijela.
Diferencijalna dijagnoza. Alergijski rinokonjuktivitis potrebno je razlikovati od ostalih oblika kao što su vazomotorni, medikamentni, infektivni i hormonski rinokonjuktivitis.
Liječenje. Osnovu liječenja čini izbjegavanje kontakta s uzročnim alergenima (uklanjanje i izbjegavanje kućnih ljubimaca, korištenje filtera za zrak, putovanje u područja bez polinacije za vrijeme kritičnoga perioda i sl.) uz primjenu farmakoloških mjera liječenja. Temelj farmakološkoga liječenja čini nanošenje lokalnih kortikosteroida na nosnu ili očnu sluznicu s ili bez peroralne primjene antihistaminika. U težim oblicima mogu se primjenjivati i drugi lijekovi i postupci (antileukotrijeni, sistemski kortikosteroidi, specifična imunoterapija).
Intranazalni kortikosteroidi predstavljaju najučinkovitiji i najpouzdaniji način liječenja koji kod većine bolesnika dovodi do trenutnoga smirivanja simptoma, iako se kod nekih bolesnika na taj učinak mora čekati i do nekoliko tjedana. Osim pri akutnom obliku bolesti, djeluju pogodno i kod kroničnoga oblika gdje dokazano smanjuju masu hipertrofične sluznice i polipa smanjujući osjećaj začepljenosti i povećavajući njegovu prohodnost. Najčešće primjenjivani lokalni kortikosteroidi su beklometazon (42 mcg 2 x 1 potisak u svaku nosnicu), flunizolid (25 mcg 2 x 1 potisak u svaku nosnicu), budesnoid (100 mcg 2 x 1 potisak u svaku nosnicu) te flutikazon (200 mcg 2 x 1 potisak u svaku nosnicu). Primjena ovih lijekova smatra se sigurnom, može se provoditi kroz dulji period, a najčešća nuspojava primjene je epistaksa. Pri korištenju ovih pripravaka preporuča se i češće ukapavanje fiziološke otopine ili sličnih pripravaka na sluznicu nosa kako bi spriječilo isušivanje i prevenirao nastanak sekundarnih gljivičnih infekcija.
Peroralni antihistaminici dodaju se u terapiju kada se uz lokalnu primjenu kortikosteroida ne postiže zadovoljavajuća kontrola bolesti. Najčešće korišteni su triciklički antihistaminici sa selektivnim djelovanjem na periferne histaminske (H1) receptore: loratadin (1 x 10 mg/dan), desloratadin (1 x 5 mg/dan) te feksofenadin (2 x 60 mg ili 1 x 100 mg/dan). Izbjegavanjem djelovanja na centralne histaminske receptore izbjegava se sedirajući učinak. Osim pospanosti, mogu izazvati glavobolju, kserostomiju, a moguć je i razvoj tolerancije što se manifestira povratkom alergijskih simptoma. Primjena ovih lijekova ograničava se na trajanje sezone alergijskih smetnji.
Ostale terapijske mjere. Kod bolesnika sa simptomima upornim unatoč standardnoj terapiji u obzir dolazi i primjena antileukotrijena, kao što je montelukast (1 x 10 mg/dan), bilo samostalno, bilo u kombinaciji s antihistaminicima. Lokalna primjena kromolina (stabilizator membrane mastocita) manje je učinkovita, a zbog brzoga uklanjanja mora se primjenjivati češće (i do četiri puta na dan). U najtežim oblicima opravdana je i kratkotrajna sistemska primjena kortikosteroida (prednizolon 20 mg kroz pet dana), a kod nekih se primjenjuju i intramuskularni depo preparati (npr. triamkinolon 40 mg) koji imaju produljen učinak. Također, kod ovih bolesnika u obzir dolazi i specifična imunoterapija u vidu hiposenzibilizacije koja se provodi supkutanom ili sublingvalnom primjenom alergena u malim koncentracijama u određenim vremenskim intervalima s povećanjem primijenjene doze što dokazano dovodi do smanjenja ukupne vrijednosti cirkulirajućega IgE-a, kao i smanjenja potrebe za ostalim farmakološkim mjerama.
Prognoza. Iako se radi o kroničnom oboljenju, uz pridržavanje mjera izbjegavanja uzročnoga alergena te redovnoj primjeni lijekova kod većine bolesnika postiže se odgovarajuća kontrola simptoma.
URTIKARIJA I ANGIOEDEM
Definicija i epidemiologija. Urtikarija predstavlja monomorfnu dermatozu koju obilježava iznenadna pojava dobro ograničenih, edematoznih i eritematoznih površina koje nastaju uslijed edema dermisa, dok je angioedem naziv za isti proces koji zahvaća i potkožno tkivo, osobito u području mekih tkiva usana, vjeđa i jezika dovodeći do njihova karakteristično izraženoga edema. Oba procesa nastaju kao posljedica degranulacije mastocita i oslobađanja vazoaktivnih medijatora, što je kod najvećega broja oboljelih potaknuto reakcijom preosjetljivosti tipa I, ali mogu biti posredovani i drugim, nealergijskim mehanizmima. Urtikarija i angioedem često se javljaju skupa, no moguća je i pojava svakoga entiteta zasebno. Smatra se da se urtikarija i/ili angioedem razviju kod 15 do 25 % pripadnika opće populacije u nekom trenu tijekom života s najvećom učestalošću kod djece i mlađih odraslih.
Etiopatogeneza. Urtikarija i angioedem nastaju uslijed degranulacije mastocita pri čemu se oslobađaju različite vazoaktivne tvari poput histamina, serotonina, eikozanoida (leukotrijeni, prostaglandini) te raznih citokina koji dovode do povećane krvožilne propusnosti s posljedičnom ekstravazacijom proteina i tekućine te stvaranjem edema. Kada se radi o urtikariji, proces se odvija u dermisu, dok se kod angioedema radi o procesu koji se širi i u potkožno tkivo. Aktivaciju i degranulaciju mastocita odgovornu za nastanak ovih poremećaja mogu izazvati različiti podražaji i mehanizmi, a najčešće se radi o alergijskoj reakciji posredovanoj reakcijom preosjetljivosti tipa I kao odgovorom na različite alergene (nutritivni alergeni, lijekovi, otrovi kukaca i insekata). Osim toga, mogu ju uzrokovati i fizikalni podražaji (pritisak i trenje, promjene temperature, izlaganje svjetlosti), infekcije (H. pylori, hepatitis B), neurogeni stimulansi (djelovanje simpatikusa) te lijekovi (aspirin, opijati). Kod velikoga dijela bolesnika točan uzrok i mehanizam nastanaka ovih poremećaja nisu poznati pa govorimo o idiopatskoj urtikariji i angioedemu.
Klasifikacija. Osnovna podjela urtikarije i angioedema temelji se na njihovom trajanju i kliničkim obilježjima tako da razlikujemo akutnu urtikariju i angioedem koji traju do šest tjedana i kroničnu urtikariju i angioedem koji traju dulje od šest tjedana. Nova klasifikacija kroničnu urtikariju i angioedem dijeli u dvije podgrupe: kronična spontana urtikarija i angioedem (engl. chronic spontaneous urticaria, CSU) te kronična inducibilna urtikarija i angioedem (engl. chronic inducibile urticaria, CIU) (Slika 9.1).
Akutna urtikarija i angioedem najčešće su posredovani infekcijama i alergijskim reakcijama, najčešće na nutritivne alergene (orašasti plodovi, jaja, mlijeko), lijekove (penicilin) te toksine kukaca. Akutna urtikarija najčešće se pojavljuje difuzno i zahvaća veće površine kože trupa i ekstremiteta.
Kronična inducibilna urtikarija i angioedem mogu biti uzrokovani različitim nealergijskim mehanizmima kao što su pritisak (urtikarija na pritisak), hladnoća (urtikarija na hladnoću), toplina (urtikarija na toplinu), Sunčeve zrake (solarna urtikarija) ili pojačana aktivnost simpatikusa (kolinergična urtikarija). Većina fizikalnih urtikarija ima naznačen dermografizam, a on se definira kao pojava urtika na koži na mjestu izravnoga mehaničkog podraživanja (grebanje, struganje).
Kronična spontana urtikarija i angioedem danas se smatraju autoimunim poremećajem s obzirom na to da se kod većine bolesnika detektiraju funkcionalna autoantitijela na IgE ili na receptor za IgE te ovi bolesnici imaju i veću pojavnost autoimunih bolesti štitne žlijezde.
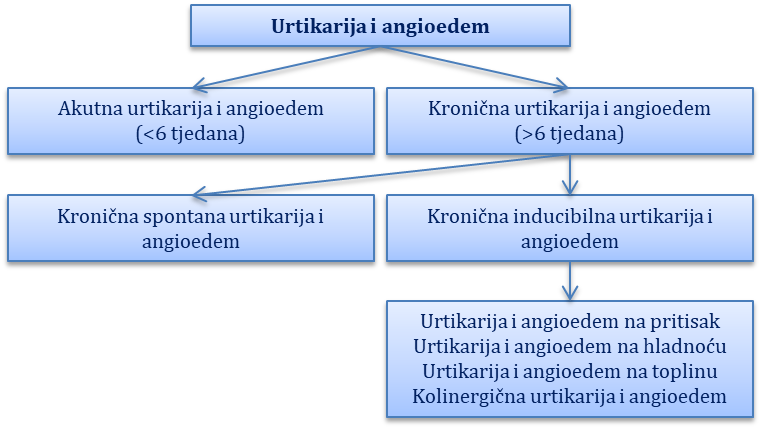
Slika 9.1. Klasifikacija urtikarije i angioedema
Klinička slika. Urtikariju karakterizira difuzna ili lokalno ograničena pojava monomorfnih kožnih promjena koje nazivamo urtikama. One označavaju oštro ograničen, induriran kožni areal koji može biti različite veličine (od nekoliko milimetara do nekoliko centimetara), a mjestimično one mogu međusobno i konfluirati. Najčešće su ružičaste do crvenkaste boje, a moguća je pojava i bijelih ili hemoragičnih urtika. Uslijed kronične urtikarije urtike su obično hiperpigmentirane. Najčešće su distribuirane po trupu i ekstremitetima, a mogu se pojaviti i na glavi i vratu. Pojava urtika praćena je osjetom svrbeža. Angioedem je praćen otokom mekih tkiva u području vjeđa, usana, jezika, genitalija te u području gornjih dišnih puteva (uvula, meko nepce, ždrijelo, grkljan) što može biti uzrok stridoroznoga disanja i kompromitiranosti dišnoga puta zbog čega ovo predstavlja potencijalno po život opasno stanje. Angioedem se najčešće nadovezuje na urtikariju, iako se može javiti i bez nje (npr. hereditarni angioedem koji je zasebno opisan). Prema trajanju opisanih simptoma razlikujemo akutnu urtikariju i angioedem (do šest tjedana), kroničnu urtikariju i angioedem (više od šest tjedana) te intermitentnu (recidivirajuću) urtikariju.
Dijagnoza se u većini slučajeva postavlja na osnovi kliničke slike, kliničkoga nalaza te anamnestičkih podataka koji su usmjereni na početak i trajanje simptoma te njihovu povezanost s nekim alergenima (hrana, piće) ili okolnostima (promjene temperature, izlaganje Suncu). Kod bolesnika s fizikalnim oblicima urtikarije pozitivan je znak dermografizma. Jasna povezanost simptoma s određenim alergenom ne zahtijeva daljnju obradu, no kod bolesnika s nejasnim uzrokom te kod recidivirajuće i kronične bolesti potrebno je provesti dijagnostičku obradu. Ona se sastoji od laboratorijskih nalaza (KKS, SE, AST, ALT, IgE, C3, C4), mikrobiološke dijagnostike (utinokultura, koprokultura, testiranje na infekciju H. pylori) te alergoloških testova s ciljem otkrivanja potencijalnoga alergena (specifični IgE, prick-test na nutritivne alergene). Kod bolesnika s kroničnom urtikarijom određuju se i specifična antitijela na IgE receptor koja su pozitivna kod velike većine bolesnika s kroničnom spontanom urtikarijom i angioedemom. U nejasnim slučajevima opravdana je i biopsija kožnih promjena.
Liječenje akutne urtikarije i angioedema sastoji se od sistemske primjene kortikosteroida i antihistaminika, obično u jednoj dozi, nakon čega slijedi peroralna (mono)terapija antihistaminicima (antagonisti histaminskih H1 receptora). Od parenteralnih kortikosteroida najčešće se primjenjuje deksametazon (4 - 8 mg iv ili im) ili metilprednizolon (40 - 125 mg iv ili im), dok se od parenteralnih antihistaminika najčešće koristi kloropiramin (20 mg iv ili im). Kod bolesnika s izraženom hipotenzijom potrebna je primjena intravenske infuzije, a kod izraženoga angioedema koji ugrožava dišni put daje se nerazrijeđeni adrenalin u dozi od 0,5 - 1 mg im ili sc. Uslijed težih oblika ponekad je potrebno ponoviti parenteralnu dozu kortikosteroida i antihistaminika i/ili nastaviti kratkotrajnu peroralnu primjenu kortikosteroida (prednizon 20 - 40 mg/dan). Nakon početne parenteralne terapije uobičajeno se liječenje nastavlja primjenom potencijalno sedativnih (loratadin 10 mg/dan, cetirizin 10 mg/dan) ili nesedativnih antagonista histaminskih H1 receptora (feksofenadin 180 mg/dan, desloratadin 5 mg/dan). Kod težih oblika moguće je povećati dozu peroralnih antihistaminika i do četiri puta više od preporučene (gore navedene) doze. Kod bolesnika kod kojih se ne može postići kontrola simptoma navedenim lijekovima moguća je primjena kombinacije antihistaminika (npr. feksofenadin i cetirizin) ili primjena antileukotrijena uz postojeći antihistaminik (montelukast 10 mg/dan; zafirlukast, 2 x 20 mg/da) ili kalcijskih blokatora (nimodipin, nefidipin) koji djeluju kao stabilizatori membrane mastocita. Peroralna terapija primjenjuje se u trajanju od pet do deset dana, odnosno do povlačenja simptoma. Uz ove mjere bolesnicima se preporuča izbjegavanje uzročnoga alergena ili okolnosti koje povezuju s pojavom simptoma, pridržavanje tzv. urtikarijske dijete (izbjegavanje orašastih plodova, mlijeka, jaja te gaziranih i obojenih pića) te izbjegavanje primjene nesteroidinih protuupalnih lijekova i opijata.
Liječenje kronične i recidivirajuće urtikarije i angioedema. Osnovu liječenja ovih bolesnika čini izbjegavanje potencijalnih alergena i/ili okolnosti koje se povezuju s nastankom simptoma, a farmakološko liječenje temelji se na dugotrajnijoj primjeni antagonista histaminskih H1 receptora (loratadin, cetirizin, feksofenadin, desloratadin) s ili bez dodatka antileukotrijena (montelukast, zafirlukast). Kod bolesnika kod kojih se ne postiže zadovoljavajuća kontrola simptoma moguće je primijeniti i antagonist histaminskih H2 receptora (ranitidin, cimetidin), kalcijski blokator (nimodipin, nifedipin) ili doksepin (triciklički antidepresiv s antihistaminskim učinkom). U najtežim oblicima indicirana je primjena glukokortikoida (prednizon 20 mg svaki drugi dan kroz tri tjedna, a potom smanjivati dozu za 2,5 do 5 mg svaka 2 do 3 tjedna) ili bioloških lijekova (omalizumab, monoklonsko antitijelo na receptor za IgE), a u rezistentnim oblicima u obzir dolazi uvođenje imunomodulatora (npr. ciklosporin 3 - 5 mg/kg/dan).
Prognoza. Akutna urtikarija i angioedem obično prolaze kroz nekoliko dana do nekoliko tjedana. Kod više od polovine bolesnika koji imaju simptome dulje od šest tjedana oni perzistiraju godinama. Uz pridržavanje općih mjera i farmakološkoga liječenja kod većine bolesnika postiže se odgovarajuća kontrola simptoma.
ANAFILAKSIJA I ANAFILAKTIČKI ŠOK
Definicija. Anafilaksija predstavlja generaliziranu, po život opasnu reakciju preosjetljivosti koju karakterizira ugroženost dišnoga puta i disanja (edem mekih tkiva, bronhoopstrukcija) te poremećaj cirkulacije u vidu razvoja distributivnoga šoka (tzv. anafilaktički šok). Iako se kod najvećega broja bolesnika radi o najtežem obliku alergijske reakcije posredovane imunoglobulinom E, ona može biti izazvana i drugim imunološki posredovanim, kao i neimunološkim mehanizmima. Procijenjena godišnja incidencija anafilaksije i anafilaktičkoga šoka relativno je visoka i kreće se od 10 do 100 slučajeva na 100 000 stanovnika. Najčešći uzroci anafilaksije u dječjoj dobi su nutritivni alergeni, dok su u odrasloj dobi najčešći uzročnici lijekovi.
Etiopatogeneza. Anafilaksija i anafilaktički šok nastaju kao posljedica aktivacije i degranulacije mastocita i bazofila pri čemu se oslobađaju različiti proupalni medijatori koji su zaslužni za razvoj sistemske reakcije u vidu distributivnoga šoka i drugih posljedica, u prvom redu edema i ugroženosti gornjih dišnih puteva, bronhoopstrukcije te kožnih promjena (urtikarija). Najvažniji medijator anafilaksije je histamin koji svoje djelovanje ostvaruje preko histaminskih receptora koji se dijele u dvije skupine (H1 i H2). Crvenilo, hipotenzija i vazodilatacija nastaju posredovanjem i H1 i H2 receptora, dok su tahikardija, svrbež, rinoreja i bronhospazam posredovani H2 receptorima. Bitan medijator je i tripraza, enzim koji ima sposobnost aktivacije sustava komplementa te kalikrein-kininskoga sustava, što za posljedicu ima nastanak angioedema i hipotenzije. Ostali medijatori u anafilaksiji i njihovi učinci prikazani su u Tablici 9.2. Navedena aktivacija i degranulacija mastocita i bazofila može biti uzrokovana različitim imunološkim i neimunološkim mehanizmima.
|
Tablica 9.2. Medijatori anafilaksije i njihovi učinci
|
|
Medijator anafilaksije
|
Učinak
|
|
Histamin
|
Povećana propusnost krvnih žila, kontrakcije glatke muskulature, hipersekrecija
|
|
Serotonin
|
Povećana propusnost krvnih žila, stimulacija kontrakcije glatke muskulature
|
|
Kemotaksijski faktor ECF-A
|
Kemotaksija eozinofila
|
|
Faktor agregacije trombocita
|
Nakupljanje i degranulacija trombocita, kontrakcija dišne glatke muskulature
|
|
Leukotrijeni
|
Povećana propusnost krvnih žila, kontrakcija dišne glatke muskulature
|
|
Kinini
|
Povećana propusnost krvnih žila, kontrakcija glatke muskulature
|
|
Prostaglandini
|
Vazodilatacija, kontrakcija glatke muskulature, nakupljanje trombocita
|
Imunološki mehanizmi razvoja anafilaksije. Najčešći patofiziološki mehanizam nastanka anafilaksije predstavlja alergijsku reakciju posredovanu imunoglobulinom E (reakcija preosjetljivosti tip I ili alergijsko-anafilaktički tip). U ovom obliku okidač predstavlja alergen koji dolazi u interakciju s već stvorenim IgE-om koji se nalazi na površini mastocita i bazofila kod prethodno senzibiliziranih osoba, a posljedično ovoj interakciji dolazi do aktivacije unutarstaničnih puteva koji će završiti aktivacijom i degranulacijom navedenih stanica. Najčešći alergeni koji dovode do anafilaksije su: hrana (kiririki, školjke i ribe, mlijeko, jaja, soja, voće), lijekovi te ubodi (otrovi) kukaca. Anafilaksija može biti posredovana i imunološkim mehanizmima koji nisu ovisni o IgE-u (reakcija posredovana IgG protutijelima ili imunokompleksima), najčešće su anafilaktičke reakcije na radioaktivni kontrast, lijekove (nesteroidni protuupalni lijekovi), dekstrane i biološke agente (monoklonalna antitijela).
Neimunološki mehanizmi razvoja anafilaksije. Anafilaksija može nastati i direktnom aktivacijom mastocita i bazofila bez utjecaja imunoloških mehanizama, što se često naziva anafilaktoidnom reakcijom ili pseudoanafilaksijom. Okidači koji mogu dovesti do ovih reakcija su različiti fizikalni i kemijski stimulansi koji mogu uzrokovati aktivaciju receptora za IgE na površni mastocita i bazofila. Najčešći fizikalni stimulansi su tjelesna aktivnost, pothlađivanje te izlaganje Sunčevoj svjetlosti, dok su etanol i neki lijekovi (opioidi) najčešći primjeri kemijskih stimulansa. Najpoznatija i najučestalija anafilaktoidna reakcija je ona nakon primjene acetilsalicilne kiseline i drugih NSAID-a. Smatra se da je temeljni mehanizam ove reakcije skretanje metabolizma arahidonske kiseline s puta ciklooksigenaze, koja je blokirana ovim lijekom, na lipooksigenazu, posljedica čega je stvaranje veće količine leukotrijena koji imaju anafilaktoidne učinke. U ovu skupinu ubrajamo i tzv. idiopatsku anafilaksiju kojoj se ne može dokazati (otkriti) potencijalni okidač (ranije neprepoznati alergeni, poremećaj mastocita).
Čimbenici koji pridonose razvoju anafilaksije su dob (ugrožene skupine su djeca, adolescenti te starija populacija), pridružene bolesti (anafilaksija, mastocitoza, atopijska oboljenja), primjena određenih lijekova (beta blokatori, ACE inhibitori), konzumiranje alkohola te prisutnost čimbenika koji pojačavaju anafilaktoidni odgovor (tjelesna aktivnost, akutna infekcija, emocionalni stres te prijemenstruacijski period kod žena).
Klinička slika. Kliničke manifestacije anafilaksije nastaju relativno brzo nakon kontakta s potencijalnim okidačem, obično kroz nekoliko minuta do nekoliko sati (proteinski antigeni brže izazivaju anafilaksiju u odnosu na ugljikohidratne). Osnovni simptomi i znakovi koji obilježavaju anafilaksiju i anafilaktički šok navedeni su u tablici, slijedom ABCDE pristupa životno ugroženom bolesniku (Tablica 9.3). Prekid simptoma uglavnom je izazvan primjenom lijekova, a kod manjega broja oboljelih moguće je i spontano prolaženje uslijed aktivacije kompenzacijskih mehanizama (endogeni adrenalin, angiotenzin II, endotelin). Kod 5 do 20 % oboljelih nalazimo bifazični tijek bolesti koji označava ponovni povrat simptoma, obično se zbiva unutar prvih osam sati od povlačenja simptoma, iako su opisani slučajevi povrata simptoma i nakon 30 sati. Rekurentni simptomi obično su blaži od inicijalnih, najčešće u obliku urtikarije. Rizik od bifazičnoga tijeka bolesti imaju bolesnici s inicijalno teškom epizodom, bolesnici koji su zahtijevali primjenu dvostruke doze adrenalina, bolesnici s protrahiranom hipotenzijom te bolesnici koji su se duže oporavljali od inicijalne epizode.
|
Tablica 9.3. Klinička slika anafilaksije i anafilaktičkoga šoka
|
|
ABCDE pristup
|
Karakteristike u anafilaksiji i anafilaktičkom šoku
|
|
A (airway) – dišni put
|
Edem mekih tkiva (uvula, ždrijelo, jezik), promuklost, inspiratorni stridor
|
|
B (breathing) – disanje
|
Dispneja, bronhospazam, hipoksična konfuzija, respiratorni arest
|
|
C (circulation) – cirkulacija
|
Blijeda i oznojena koža, tahikardija, hipotenzija, bradikardija, kardijalni arest
|
|
D (disability) – neurološka procjena
|
Poremećaj stanja svijesti (somnolencija, sopor, koma)
|
|
E (exposure) – izloženost
|
Eritem, urtikarija, angioedem
|
Dijagnoza anafilaksije i anafilaktičkoga šoka temelji se na kliničkoj slici, kliničkom pregledu te anamnestičkim podacima (izloženost alergenu ili okolnostima koje pogoduju nastanku anafilaksije). Dijagnostički kriteriji za anafilaksiju i anafilaktički šok navedeni su u Tablici 9.4. Nakon inicijalnoga zbrinjavanja ovih bolesnika daljnja dijagnostička obrada identična je kao i kod urtikarije i angioedema.
|
Tablica 9.4. Dijagnostički kriteriji za anfilaksiju/anafilaktički šok
|
|
Anafilaksija i anafilaktički šok vrlo su vjerojatni kada je zadovoljen jedan od tri navedena kriterija:
|
|
Kriterij 1
|
Akutni početak bolesti (od jedne minute do nekoliko sati) s vidljivim promjenama na koži i/ili sluznicama (urtikarija, svrbež ili crvenilo, otok usana, jezika ili uvule) uz barem jedan od navedenih kriterija:
a) poremećaj disanja (dispneja, bronhospazam, stridor, hipoksemija),
b) hipotenzija ili odgovarajući simptomi potpune disfunkcije organa (sinkopa, kolaps i sl.).
|
|
Kriterij 2
|
Dva ili više odgovarajućih simptoma koji slijede vrlo brzo nakon izlaganja vjerojatnom alergenu (od jedne minute do nekoliko sati):
a) promjene na koži i/ili sluznicama (urtikarija, angioedem),
b) poremećaj disanja (dispneja, bronhospazam, stridor),
c) hipotenzija ili odgovarajući simptomi potpune disfunkcije organa (sinkopa, kolaps i sl.),
d) stalni gastrointestinalni simptomi (grčevi, povraćanje, proljev).
|
|
Kriterij 3
|
Snižen krvni tlak nakon izlaganja poznatom alergenu kod pacijenta (od jedne minute do nekoliko sati):
a) dojenčad i djeca: niski sistolički krvni tlak (dobna specifičnost) ili pad sistoličkoga tlaka veći od 30 %,
b) odrasli: sistolički krvni tlak niži od 90 mm Hg ili pad veći od 30 % normalnoga tlaka osobe.
|
Liječenje bolesnika s anafilaksijom i anafilaktičkim šokom ne smije se odgađati, mora biti promptno s obzirom na brz razvoj po život opasnih komplikacija (kardijalni ili respiratorni arest). Bolesnika treba staviti u odgovarajući položaj (sjedeći kod dominacije respiratornih tegoba, odnosno ležeći ili Trendeleburgov položaj kod dominacije hemodinamske nestabilnosti), osigurati odgovarajuću oksigenoterapiju te otvoriti venski put kako bi se mogla primijeniti parenteralna terapija. U slučaju razvoja kardiorespiratornoga aresta potrebno je odmah započeti s mjerama kardiopulmonalne reanimacije. Jedan od najvažnijih koraka u postupku zbrinjavanja ovih bolesnika je i zaustaviti djelovanje tvari koja je dovela do anafilaksije (najčešće se radi o prekidu davanja lijeka).
Osiguranje dišnoga puta jedna je od osnovnih zadaća u zbrinjavanju ovih bolesnika. Kada se procijeni da se radi o ugroženosti dišnoga puta ili o brzom razvoju simptoma i znakova anafilaksije koji bi mogli završiti ugrožavanjem prohodnosti dišnoga puta, indicirana je endotrahealna intubacija, a ako je ona nemoguća, i hitna konikotomija ili traheotomija.
Adrenalin je osnovni i lijek izbora u liječenju ovih bolesnika te bi ga trebale primiti sve životno ugrožene osobe. Osnovni put primjene adrenalina pri anafilaksiji i anafilaktičkom šoku je intramuskularni (anteromedijalni dio natkoljenice), i to u dozi od 0,5 mg, odnosno 0,5 ml za odrasle osobe (nerazrijeđeni adrenalin dolazi u ampulama od 1 ml u kojima je po 1 mg adrenalina). Prema potrebi se doza ponavlja svakih pet minuta. Adrenalin ostvaruje djelovanje posredstvom alfa (vazokonstrikcija) te beta adrenergičkih receptora (pojačana kontraktilnost, bronhodilatacija). Supkutana i inhalacijska primjena adrenalina više se ne preporuča, a intravenska primjena opravdana je samo od strane iskusnih liječnika (u bolusima od 50 mcg). Bolesnici s preboljenim anafilaktičkim šokom trebaju posjedovati adrenalinske autoinjektore koje mogu sami primijeniti u slučaju ponovne pojave anafilaksije.
Ostale farmakološke mjere u liječenju anafilaksije i anafilaktičkoga šoka uključuju odgovarajuću volumnu resuscitaciju (500 - 1000 ml 0,9 %-tnoga NaCl u bolusu), primjenu antagonista histaminskih H1 receptora (kloropiamin 20 mg) te H2 receptora (ranitidin 50 mg), kortikosteroida (hidrokortizon 200 mg, metilprednizolon 125 mg). Prema potrebi se primjenjuju i bronhodilatatori (inhalacije slabutamola i/ili ipratropija, aminofilin), glukagon (kod liječenja anafilaksije bolesnika koji uzimaju beta blokatore) te kontinuirana infuzija noradrenalina za ispravak hipotenzije.
Nakon stabilizacije bolesnika liječenje se provodi kao i kod urtikarije i angioedema. Bolesnici s teškom kliničkom slikom anafilaksije, kao i oni koji su zahtijevali opetovanu primjenu adrenalina, zahtijevaju hospitalizaciju i promatranje kroz minimalno 24 sata, a produljeni nadzor preporuča se za bolesnike kod kojih je reakcija nastala nakon 30 minuta od kontakta s potencijalnim okidačem ili je prošlo više od 60 minuta od pojave simptoma do primjene adrenalina.
Prognoza. Smrtnost od anafilaksije u današnjim uvjetima manja je od 1 %. Rizik od smrtnoga ishoda veći je što je brže nakon kontakta s potencijalnim uzrokom reakcija nastala. Smrtni slučaj nikada nije zabilježen u slučaju reakcije koja je nastala nakon 6 sati od izloženosti potencijalnom uzroku anafilaksije.
Definicija. Hereditarni angioedem je rijetka, potencijalno za život opasna bolest koju karakteriziraju nepredvidivi napadaji lokaliziranih, asimetričnih edema udova, lica, trupa, probavnoga ili dišnog sustava, koji mogu dovesti do teških poremećaja koji izravno ugrožavaju život bolesnika. Najpoznatiji oblik bolesti nastaje kao posljedica nasljednoga poremećaja gena koji kodira inhibitor C1 komponente komplementa (tzv. C1 esetaraza). U nedostatku toga inhibitora dolazi do spontane aktivacije sustava komplementa čime se aktiviraju i komponente koje imaju anafilaktoidne učinke, najzaslužnije za kliničku prezentaciju ove bolesti.
Epidemiologija. Hereditarni angioedem je rijetka bolest te čini oko 2 % svih angioedema i ima incidenciju oko 1 na 50 do 100 tisuća ljudi u općoj populaciji. Etnička pripadnost i spol ne utječu na pojavnost bolesti, iako je primjećeno da su žene sklonije težim kliničkim prezentacijama nego muškarci. Iako je deficit C1 esteraze prisutan već po rođenju, rijetko se bolest prezentira u perinatalnom periodu. Kod većine oboljelih prvi napadaju se javljaju u prvom ili drugom desetljeću života.
Etiologija. Hereditarni angioedem najčešće nastaje kao posljedica genetske mutacije gena za C1 inhibitor, koja može nastati spontano (de novo) ili se prenosi autosomno-dominanatno. Također, hereditarni angioedem može biti posljedica i nekih stečenih bolesti i stanja.
Genetski oblik hereditarnoga angioedema. Gen za C1 esterazu (SERPING1) nalazi se na 11. kromosomu, a do danas je opisano više od 300 mutacija koje se povezuju s nastankom ovoga poremećaja. U 25 % slučajeva radi se o novonastaloj mutaciji, dok se u ostalim slučajevima (75 %) radi o autosomno-dominantnom nasljeđivanju. Na taj način nasljeđivanja ukazuje činjenica da nema zdravih nositelja genetskoga defekta. Zbog izuzetne rijetkosti bolesti, svi oboljeli su heterozigoti jer mogućnost da oba roditelja boluju od hereditarnoga angioedema (što je preduvjet za nastanak homozigota i postojanje dva defektna gena) gotovo je nemoguća. Sinteza proteina blokirana je na strani mutiranoga alela, a održana na strani normalnoga alela te o penetrantnosti mutiranoga gena ovisi i klinička prezentacija bolesti (asimptomatski oblik, teški oblik).
Stečeni oblik hereditarnoga angioedema. Osim nasljedne komponente bolesti, danas se sve više govori i o stečenom obliku manjka C1 esteraze što je povezano s limfoproliferativnim bolestima (uslijed pojačane potrošnje komplementa), autoimunim poremećajima (razvoj autoantitijela na C1 inhibitor) te nekim lijekovima, poglavito ACE inhibitorima (uslijed pojačanoga nakupljanja bradikinina).
Patogeneza. Uslijed genetskoga defekta dolazi do smanjenje sinteze C1 inhibitora ili je ona očuvana, ali se radi o nefunkcionalnom proteinu. Kao posljedica toga izostaje inhibicija komponente C1 te dolazi do spontane aktivacije sustava komplementa čime se oslobađaju vazoaktivne tvari koje dovode do nastanka edema. Nastanku edema također pogoduje i stvaranje veće količine bradikinina iz kalikreina, s obzirom na to da C1 inhibitor inhibira i to pretvaranje. Bradikinin, kao i određene komponente komplementa (C3a, C5a, C4a), imaju vazoaktivno djelovanje tako što dovode do vazodilatacije i povećane permeabilnosti kapilara zbog čega dolazi do izlaska tekućine u intersticijski prostor (ekstravazacija) i nastanka edema kože, potkože i sluznica.
Klasifikacija. S obzirom na etiološku osnovu, razlikujemo dva oblika hereditarnoga angioedema: hereditarni angioedem koji nastaje kao posljedica snižene razine ili smanjene funkcije plazmatskoga C1 inhibitora (HAE-C1-INH) kojima ima dva podtipa (tip I i tip II) te hereditarni angioedem s normalnom razinom i funkcijom plazmatskoga C1 inhibitora (HAE-nl-C1-INH).
HAE-C1-INH tip I najčešći je oblik (85 %) hereditarnoga angioedema, a obilježen je sniženom razinom C1 inhibitora što je posljedica njegove smanjene sinteze. Iako je zahvaćen samo jedan alel, drugi alel nije dostatan za sintezu količine C1 inhibitora u dovoljnoj količini da bi spriječio aktivaciju sustava komplementa i napad angioedema.
HAE-C1-INH tip II obilježen je sintezom nefunkcionalnoga C1 inhibitora, dok je njegova koncentracija u plazmi normalna ili čak povišena. Ovaj oblik najčešće je posljedica spontane mutacije, a rjeđe se radi o nasljednom poremećaju. Čini oko 15 % hereditarnih angioedema.
HAE-nl-C1-INH tip (ranije zvan kao tip III) predominantan je kod žena, a klinički može nalikovati na HAE-C1-INH, a razlikuje ga odsutnost pojave eritema marginatu te dulji vremenski period između dva napadaja (napadaji se pojavljuju rjeđe). Ovaj oblik najčešće nastaje kao posljedica mutacije faktora XII, zatim angiopietin-1, plazminogena ili kininogen-1 teškoga lanca, ali također se može raditi i o novoj, nepoznatoj mutaciji. Najčešće se javlja nakon djetinjstva, a anamnestički podatak o angioedemu u obitelji ključan je za postavljanje dijagnoze. Od svih oblika HAE-a, ovaj se smatra u najvećoj mjeri uvjetovanim estrogenom te je često provociran trudnoćom i uzimanjem oralnih kontraceptiva.
Klinička slika. Kod bolesnika s HAE-C1-INH tip I i II napadaji započinju već u djetinjstvu, dok se kod HAE-nl-C1-INH uglavnom javljaju kasnije, u mlađoj i srednjoj životnoj dobi. Bolest karakteriziraju napadaji (atake) između kojih nalazimo periode remisije u kojima se bolesnici osjećaju dobro i nemaju simptoma. Učestalost pojave napadaja hereditarnoga angioedema je varijabilna, tako da se kod nekih oboljelih javlja jednom tijekom života, dok se kod drugih napadaji javljaju na tjednoj bazi. Iako nema točne poveznice, napadajima često prethode određeni okidači kao što su trauma, veći tjelesni ili emocionalni napor, virusne ili bakterijske infekcije, menstruacija kod žena i slično.
Velik dio bolesnika navodi prodromalne simptome prije pojave napada hereditarnoga edema. Najčešće su to ispadi osjeta u vidu parestezija na području kože gdje će se razviti lokalizirani edem, osjećaj suhoće i grebanja u grlu te promuklost prije edema gornjih dišnih puteva, kao i opća slabost, malaksalost te promjene raspoloženja. Prodromalni simptomi obično se javljaju sat do dva prije nego se bolest počne manifestirati u svojoj punoj slici.
Napadaj hereditarnoga angioedema može se očitovati pojavom edema na jednom ili više anatomskih lokalizacija, a najčešće su to: koža i potkožno tkivo, gornji dišni putevi te abdominalni organi. Danas se sve više govori i o nastanku moždanoga edema i neurološkoj simptomatologiji kao dijelu slike hereditarnoga angioedema. Edemi u hereditarnom angioedemu nastaju i pojačavaju se tijekom 12 do 24 sata, a potom postupno i spontano regrediraju, a to nekada može trajati i do pet dana. Najčešće se povlače za 1 do 3 dana.
Promjene na koži i potkožnom tkivu najčešće su, a ujedno i najkarakterističnije značajke hereditarnoga angioedema. Najčešće zahvaćaju lice i vrat, potom trup i ekstemitete te rjeđe genitalije. Radi se o bezbolnim, blijedim, ograničenim, asimetričnim edemima koji nisu povezani s urtikarijom, svrbežom i ne prolaze nakon primjene antihistaminika. Kod jednoga dijela bolesnika na području kože iznad edema može se javiti obrubljeno crvenilo (eritema marginatum). Sat do dva prije pojave edema bolesnici mogu osjetiti parestezije u vidu trnaca i žarenja na zahvaćenom području.
Edem u području gornjih dišnih puteva najteža je i najopasnija manifestacija hereditarnoga edema, a najčešće zahvaća jezik i grkljan. Može napredovati do potpune opstrukcije što će zahtijevati hitno zbrinjavanje dišnoga puta (hitna endotrahealna intubacija, perkutana dilatativna traheostomija). Bolesnici najprije osjećaju trnce i grebanje u grlu, postaju promukli, a potom se razvija stridorozno disanje kako opstrukcija napreduje. Kod potpune opstrukcije bolesnik više ne može disati što predstavlja glavni uzrok smrti tijekom napada hereditarnoga angioedema. Smatra se da oko 70 % osoba s hereditarnim angioedemom bar jednom tijekom života ima napadaj koji se manifestira edemom larinksa i glotisa.
Abdominalne manifestacije hereditarnoga angioedema nastaju kao posljedica edema sluznice organa probavne cijevi, rjeđe mokraćnih puteva. Kod zahvaćanja organa probavne cijevi dominiraju abdominalne kolike, mučnina, povraćanje, proljev, a kod potpune opstrukcije i prekida pasaže nastaje opstipacija. Simptomi, kada su izraženi, vjerno oponašaju sliku kirurškoga akutnog abdomena i vrlo često se takvi bolesnici podvrgavaju hitnim abdominalnim operacijama. Afekcija organa mokraćne cijevi očituje se suprapubičnom boli te dizuričnim tegobama, a zbog uskoga lumena vrlo brzo se razvija anurija i različit stupanj opstruktivne uropatije.
Fizikalni pregled u fazi remisije uglavnom je uredan i ne nalazimo specifične stigme bolesti. Nalaz tijekom napadaja ovisi o širini manifestacije napadaja hereditarnoga angioedema, tako da se mogu naći: bljedoća kože i sluznica, lokalizirani edemi kože i potkože s eventualno razvijenim marginalnim eritemom različite distribucije, hipotenzija zbog ekstravazacije tekućine, tahikardija kao kompenzacijski mehanizam, tahidispneja i stridor kod opstrukcije gornjih dišnih puteva s eventualnim razvojem centralne cijanoze, trbuh miže biti meteorističan, palpatorno bolan s razvojem ascitesa u najtežim oblicima i sl.
Dijagnoza. Glavni korak u postavljanju dijagnoze hereditarnoga angioedema leži u samoj sumnji na njezino postojanje. Pri tome se najviše treba osloniti na anamnestičke podatke o samom toku bolesti, kao i o postojanju angioedema u obitelji te fizikalnoga nalaza pri samome napadu. Kada se jednom postavi sumnja na hereditarni angioedem, dijagnoza se postavlja laboratorijskim određivanjem specifičnih komponenata komplementa. Dijagnoza hereditarnoga angioedema, kao i njegova klasifikacija na podtipove, postavlja se određivanjem komponenata komplementa. Standardno se određuju sljedeće komponente komplementa: koncentracija C1 inhibitora, funkcija C1 inhibitora, koncentracija C4 te koncentracija C4q (Tablica 9.5).
|
Tablica 9.5. Određivanje serumske koncentracije pojedinih komponenti komplementa u dijagnostici HAE-a
|
|
Komponente komplementa
|
HAE-C1-INH tip I
|
HEA-C1-INH tip II
|
HEA-nl-C1-INH
|
Idiopatski
angioedem
|
Stečeni HEA
|
Inducirani
ACE-inibitorom
|
|
Koncentracija C1 inhibitora
|
Niska
|
Normalna/ visoka
|
Normalna
|
Normalna
|
Niska
|
Normalna
|
|
Funkcija C1 inhibitora
|
Slaba
|
Slaba
|
Normalna
|
Normalna
|
Slaba
|
Normalna
|
|
Koncentracija komponente C4
|
Niska
|
Niska
|
Normalna
|
Normalna
|
Niska
|
Normalna
|
|
Koncentracija komponente C1q
|
Normalna
|
Normalna
|
Normalna
|
Normalna
|
Niska
|
Normalna
|
Kod bolesnika kod kojih se dokaže hereditarni angioedem ponavljanje određivanja koncentracije komplemenata nije potrebno. Također, mjerenje komponente C4 pri napadaju hereditarnoga angioedema potvrđuje dijagnozu. Normalna vrijednost C4 za vrijeme akutnoga napadaja hereditarnoga angioedema isključuje manjak C1 inhibitora.
Bolesnici s hereditarnim angioedemom imaju uglavnom normalne rutinske laboratorijske nalaze. Tijekom napada može se zabilježiti hemokoncentracija, prerenalna azotemija te leukocitoza. Kod bolesnika koji se prezentiraju slikom akutnoga abdomena potrebno je standardnim pretragama isključiti druge uzroke (ultrazvuk trbuha, nativna radiografska snimka trbuha, kompjuterizirana tomografija).
Diferencijalna dijagnoza. S obzirom na to da postoji više vrsta angioedema, diferencijalna dijagnoza može biti otežana. Pri tome treba misliti na stečeni angioedem, angioedem izazvan lijekovima, alergijski angioedem, pseudoalergijski angioedem te idiopatski angioedem (Tablica 9.6.).
|
Tablica 9.6. Klasifikacija angioedema, njihovi uzroci i medijatori
|
|
Tip angioedema
|
Uzrok
|
Medijator
|
|
Hereditarni angioedem
|
Mutacija gena za C1 inhibitor
|
Bradikinin
|
|
Stečeni angioedem
|
Teške/kronične bolesti
|
Bradikinin
|
|
Angioedem izazvan lijekovima
|
ACE inhibitori, estrogeni
|
Bradikinin
|
|
Alergijski angioedem
|
Alergeni
|
Histamin
|
|
Pseudoalergijski angioedem
|
NSAID i slični lijekovi
|
Razni
|
|
Idiopatski angioedem
|
Nepoznat
|
Nepoznat
|
Liječenje. Terapijski pristup bolesniku s hereditarnim angioedemom temelji se i oslanja na revidiranim preporukama koje je donijela Svjetska alergološka organizacija i Europska akademija za kliničku imunologiju i alergologiju (EAACI) 2017. godine. U terapijskom pristupu razlikujemo liječenje akutnoga napadaja, kratkoročnu te dugoročnu profilaksu hereditarnoga angioedema. S obzirom na to da svaki napadaj hereditarnoga angioedema može biti potencijalno životno ugrožavajući, on se smatra hitnim stanjem i kao takav zahtijeva brzo zbrinjavanje bolesnika i primjenu odgovarajućih lijekova.
Kako je edem gornjega dišnoga puta najopasnija manifestacija hereditarnoga angioedema, prva intervencija kod takvih bolesnika mora biti usmjerena na zbrinjavanje dišnoga puta, tj. treba brzo procijeniti postoji li potreba za endotrahealnom intubacijom ili traheotomijom kako bi se osigurala prohodnost dišnoga puta.
Pri sumnji na hereditarni angioedem odmah treba započeti i liječenje lijekovima. Prema preporukama, prva linija medikamentnoga liječenja je primjena koncentrata C1 inhibitora te blokatora bradikininskih receptora (ikatibant) ili inhibitora kalikreina (ekalantid). Ako ti lijekovi nisu na raspolaganju, trebalo bi primijeniti plazmu obrađenu detergentom (SDP, engl. solvent detergent-treated plasma), a ako ni ona nije na raspolaganju, treba primijeniti svježe smrznutu plazmu. Zajedno s ovom terapijom može se primjenjivati i druga simptomatska terapija (analgetici, antiemetici, inhibitori protonske pumpe, infuzije, sedativi).
Koncentrat C1 inhibitora na tržištu dolazi u dva oblika: dobiven iz humane plazme ili rekombinantni. Primjena koncentrata C1 inhibitora iz ljudske krvi brzo nadomješta deficit enzima u plazmi i tako prekida aktivaciju sustava komplementa. Na europskom tržištu postoji nekoliko registriranih oblika ovoga lijeka. Poluvijek života ovih pripravaka duži je od 24 sata, prijavljen je minimalan broj nuspojava, a nema dokaza da se na ovaj način mogu prenijeti hepatitis B, C ili HIV. Mogu se primjenjivati u svim životnim dobima, kod trudnica, kao i za vrijeme dojenja. Rekombinantni pripravak C1 inhibitora ima isti mehanizam djelovanja kao C1 dobiven iz plazme. Ovakav pripravak nastaje u mliječnim žlijezdama transgenetskih zečica te se dobiva iz njihova mlijeka pa prije primjene treba isključiti alergiju na zečeve.
Ikatibant je blokator bradikininskih receptora tipa II. Radi se o kompetitivnom antagonistu bradikininskih receptora tipa II, što znači da se molekula zbog svoje sličnosti s bradikininom natječe za navedene receptore, veže se na njih i tako sprečava vezanje endogenoga bradikinina na receptor i posljedično dolazi do izostanka njegova učinka. U više od 90 % slučajeva samo jedna doza od 30 mg (3 ml) primijenjenoga supkutano brzo (oko 30-ak minuta) prekida razvoj kožnih, laringealnih i intestinalnih edema te sprečava nove napadaje. Primjenjuje se kod svih oblika hereditarnih angioedema kod odraslih. Ako jedna doza lijeka nije dovoljna za prekid simptoma napadaja, sljedeća injekcija može se ponoviti za 6 sati, a dnevno se maksimalno preporuča do tri injekcije. Nuspojave su uglavnom lokalne (eritem, edem, peckanje, svrbež). Ovaj se lijek preporuča pri automedikaciji, tj. u slučaju hitne samoprimjene lijeka.
Ekalantid je inhibitor kalikrenina čija se primjena također bazira na suspenziji nastanka bradikinina. Lijek nije odobren na europskom tržištu, a primjenjuje se u SAD-u te Kanadi. Postoje zabilježene anafilaktičke reakcije na taj lijek. Primjenjuje se u dozi do 30 mg supkutano i to osobama starijim od 16 godina.
Lanadelumab je biološki lijek, odobrila ga je 2015. godine Europska agencija za lijekove kao profilaksu za napadaje HAE-a za odrasle i djecu ≥ 12 godina. Lanadelumab je humano IgG1 monoklonalno antitijelo koje selektivno inhibira plazmatski kalikrein. Preporučena doza je 300 mg svaka dva tjedna, što se u slučaju dobro kontrolirane bolesti može produžiti na svaka četiri tjedna. LIjek se pokazao kao izuzetno učinkovit u kliničkim ispitivanjima u prevenciji ataka.
Svi bolesnici koji imaju dijagnozu hereditarnoga angioedema trebali bi imati lijekove za samoprimjenu u slučaju nastanka napadaja, kako bi se što prije spriječila progresija edema, osobito laringealnoga. U tu svrhu najčešće se koristi ikatibant.
S obzirom na to da mali kirurški zahvati, stomatološke intervencije, porođaj i slične situacije mogu biti okidači za napadaj hereditarnoga angioedema, preporuča se kratkoročna profilaksa neposredno prije takvih događaja. U tu svrhu se primjenjuje koncentrat C1 inhibitora 1 do 6 sati prije procedure, a ako nije dostupan, može se primijeniti atenuirani androgen, kakav je danazol, i to 5 dana prije i 2 do 5 dana nakon zahvata.
Dugoročna profilaksa napadaja hereditarnoga angioedema primjenjuje se kod bolesnika koji imaju učestale atake (jedna ili više težih ataka u mjesec dana) te kod bolesnika kada hitna terapija nije dostupna ili nije učinkovita. U dugoročnoj profilaksi koriste se koncentrat C1 inhibitora ili androgeni, iako ni jedan lijek ne osigurava sigurnu zaštitu od pojave napadaja hereditarnoga angioedema, te lanadelumab koji se pokazao vrlo učinkovit u prevenciji ataka. Prije dugoročne profilakse koncentratom C1 inhibitora potrebno je provesti cijepljenje protiv hepatitisa, HIV-a te humanoga T-limfotropnog virusa i parvovirusa. Također, treba biti oprezan pri dugoročnoj primjeni androgena zbog njegove hepatotoksičnosti, utjecaja na metabolički profil (hiperlipidemija), a kod žena može dovesti i do virilizacije i amenoreje.
Liječenje hereditarnoga angioedema mora biti individualizirano te mora uključivati preventivne mjere, samoprimjenu odgovarajuće terapije te plan zbrinjavanja hitnoga stanja. Bolesnici bi trebali stalno sa sobom nositi lijekove za hitnu primjenu u slučaju potrebe te identifikacijsku karticu sa uputama. Danas je glavni fokus na samoprimjeni lijeka jer odgoda liječenja do dolaska u zdravstvenu ustanovu povećava mortalitet. Samoprimjena lijeka u kućnim uvjetima odnosi se na vensku primjenu koncentrata C1 inhibitora, ili još praktičnije, supkutanu primjenu ikatibanta. U tu svrhu mora biti temeljito provedena edukacija, kako bolesnika, tako i njegovih ukućana i članova obitelji. Također, bolesnici bi trebali izbjegavati provocirajuće čimbenike kao što su traume, prejake fizičke aktivnosti, oralne kontraceptive i hormonsko nadomjesno liječenje, primjenu ACE inhibitora kao i druge faktore poput umora, stresa, febriliteta i konzumacije alkohola.
Definicija i epidemiologija. Mastocitoze su naziv za heterogenu skupinu bolesti koje karakterizira proliferacija i infiltracija mastocita u koži i/ili drugim tkivima i organima. Radi se o skupini rijetkih bolesti čija točna incidencija i prevalencija nisu utvrđene, a zna se da se mogu javiti u bilo kojoj životnoj dobi s nešto češćom pojavnošću u dječjoj dobi i adolescenciji.
Etiopatogeneza. Glavna patogenetska značajka mastocitoza je pojačano nakupljanje mastocita unutar određenih tkiva i organa gdje oni mogu biti aktivirani različitim podražajima (fizički dodir, tjelesni napor, emocionalni stres, alkohol, nesteroidni protuupalni lijekovi, opioidi, otrovi kukaca i insekata, alergeni iz hrane) s posljedičnom aktivacijom i degranulacijom medijatora poput histamina, serotonina, leukotrijena i drugih (detaljno opisani u poglavlju Anafilaksija i anfilaktički šok). Točan uzrok nakupljanja povećanoga broja mastocita nije poznat, ali se vjeruje da ono nastaje ili zbog njihove povećane proizvodnje i proliferacije ili zbog poremećaja procesa apoptoze. Do sada je najviše istražena mutacija c-KIT gena koji je odgovoran za nastanak transmembranskoga proteina mastocita čija unutarnja domena ima tirozin-kinaznu aktivnost, a vanjska djeluje kao receptor za faktore rasta. Mutacije ovoga gena mogu biti povezane s klonalnom ekspanzijom i poremećajem apoptoze mastocita te posljedično, njihovim nakupljanjem i infiltracijom različitih tkiva i organa (sistemska mastocitoza). Navedena mutacija je obično stečena i nije opažena obiteljska pojavnost i nasljeđivanje poremećaja. Također, istraživane su i mutacije drugih gena kao što su TET2, SRSF2, ASXL2, CBL i RUNX1 u nastanku ovih poremećaja.
Klasifikacija. Prema Svjetskoj zdravstvenoj organizaciji razlikujemo sedam temeljnih oblika mastocitoza koji se temelje na njihovim kliničkim obilježjima i pojavljivanju: 1) kutana (kožna) mastocitoza, 2) indolentna sistemska mastocitoza, 3) sistemska mastocitoza uduružena s klonalnom hematološkom bolesti nemastocistne loze, 4) agresivna sistemska mastocitoza, 5) mastocitna leukemija, 6) mastocitni sarkom te 7) ekstrakutani mastocitom.
Klinička slika. U kliničkoj prezentaciji mastocitoza razlikujemo lokalizirane (kožne) i sistemske manifestacije. Pojava simptoma i znakova je epizodična, a može biti izazvana navedenim potencijalnim podražajima, iako se mogu javiti i spontano.
Kožna mastocitoza obično se pojavljuje u dječjoj dobi, najčešće u obliku pigmentirane urtikarije (urtike poprimaju žućkasto-ružičastu ili smeđu boju) koja se može javiti u ograničenom i difuznom obliku. Također, kožna mastocitoza može se pojaviti i u obliku kožnoga mastocitoma koji se pojavljuje u obliku čvrste papule ili manjih pločastih infiltrata koji imaju površinu nalik na koru naranče. Kod većine bolesnika s kožnom mastocitozom prisutan je Darierov znak (pojava urtikarije ili eritema nakon nježnoga dodira ili laganoga trljanja kože). Pri razvoju ovih simptoma moguća je pojava i sistemskih očitovanja, kao što su hipotenzija, grčevi u trbuhu, proljev i povraćanje, kao posljedica sistemskoga učinka otpuštenih medijatora iz mastocita.
Sistemska mastocitoza može biti praćena raznolikom kliničkom slikom koja ovisi o zahvaćenosti i distribuciji bolesti. Klasične manifestacije podrazumijevaju iznenadnu pojavu vrućine, crvenila kože i konjuktiva, grčeve u trbuhu, proljev i povraćanje te tahiakardiju i hipotenziju. Kod izražene hipotenzije moguća je i sinkopa zbog prolazne moždane hipoperfuzije. Za razliku od anafilaksije, angioedem i urtikarija nisu karakteristični za sistemsku mastocitozu. Napadaj obično traje od 30 minuta do nekoliko sati te može biti praćen fatalnim ishodom, osobito kod bolesnika s od ranije poznatim kroničnim srčanim ili plućnim bolestima.
Ostale kliničke manifestacije. Posebne kliničke manfestacije dodatno se mogu javiti zbog zahvaćenosti probavnoga sustava (hipersekrecija želučane kiseline posredovana histaminom i, kao posljedica, peptičke lezije jednjaka, želuca i dvanaesnika te edem sluznice crijeva i posljedična malapsorpcija), koštano-mišićnoga sustava (fibromialgija, ubrzana osteoporoza, patološki prijelomi) te krvi i krvotvornoga sustava (pancitopenija, eozinofilija, povezanost s drugim mijeloproliferativnim i limfoproliferativnim bolestima).
Dijagnoza se postavlja na osnovi kliničke slike i nalaza (pozitivan Darierov znak patognomoničan je za sistemsku mastocitozu), anamnestičkih podataka (povezivanje simptoma s potencijalnim podražajima), a potvrđuje se patohistološkim pregledom zahvaćene kože ili drugih tkiva, najčešće sluznice crijeva ili koštanoga tkiva. Karakterističan histološki nalaz ukazuje na povećano nakupljanje mastocita u supkutanim i perivaskularnim prostorima te pojava tzv. MEL lezija, staničnih nakupina koje se sastoje od mastocita, eozinofila i limfocita. Laboratorijska dijagnostika koja se provodi kod ovih bolesnika usmjerena je na isključivanje bolesti sa sličnim kliničkim očitovanjima (vrijednosti gastrina za isključenje Zollinger–Ellisonova sindroma, vrijednosti hidroksi-indoloctene kiseline za isključenje karcinoida i sl.). Određivanje vrijednosti pojedinih medijatora mastocita u serumu ili urinu (npr. triptaze) ili određivanje mutacija c-KIT gena izvodi se vrlo rijetko. Dijagnostički kriteriji za postavljanje dijagnoze sistemske mastocitoze prikazani su u Tablici 9.7., a o sistemskoj mastocitozi govorimo ako su prisutni jedan veliki i jedan mali kriteriji ili su prisutna tri mala kriterija.
|
Tablica 9.7. Dijagnostički kriteriji za sistemsku mastocitozu prema Svjetskoj zdravstvenoj organizaciji
|
|
Veliki dijagnostički kriteriji (major)
|
|
1) Multifokalni gusti infiltrati mastocita u koštanoj srži ili drugim ekstrakutanim organima (15 mastocita u nakupini).
|
|
Mali dijagnostički kriteriji (minor)
|
|
1) Mastociti u koštanoj srži su poremećene morfologije (> 25 %);
2) Prisutnost D816V mutacije kodona 816 na c-KIT genu;
3) Mastociti u koštanoj srži izražavaju CD2 i/ili CD25 biljege;
4) Vrijednost serumske triptaze veća je od 20 ng/mL.
|
Određivanje pojedine vrste sistemske mastocitoze prema klasifikaciji Svjetske zdravstvene organizacije temelji se na određivanju morfologije mastocita u perifernoj krvi i koštanoj srži prema dolje shematski prikazanom algoritmu (Slika 9.2).
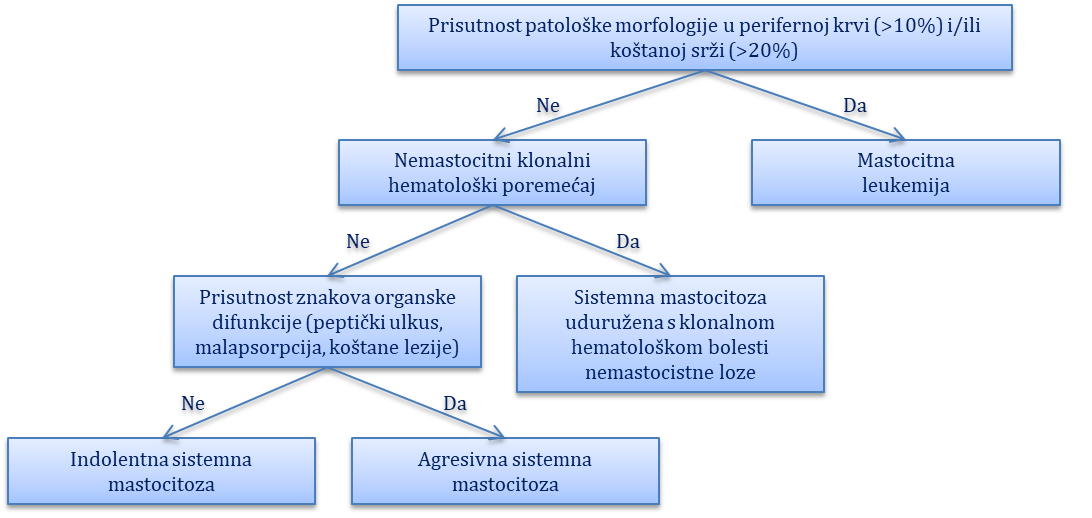
Slika 9.2. Diferencijalna dijagnoza sistemske mastocitoze
Liječenje mastocitoze ovisi o njezinoj vrsti. Liječenje kožne i indolentne sistemske mastocitoze temelji se na kontroli simptoma, što se postiže primjenom antihistaminika (feksofenadin, cetirizin, loratadin) s ili bez fotokemoterapije koja se postiže primjenom ultraljubičastih A-zraka nakon ingestije psoralena. U liječenju probavnih tegoba u prvom redu se koriste antagonisti histaminskih H2 receptora (ranitidin), a kod težih oblika dodatno se koriste i inhibitori protonske pumpe. Natrij kromolin može biti učinkovit i u smanjenju grčeva u trbuhu, proljeva, mučnine i povraćanja. U sluačaju težih kliničkih manifestacija u obzir dolazi i primjena glukokortikoida u nižim dozama te antileukotrijena. Citoreduktivna terapija (kemoterapija) rezervirana je za agresivne oblike sistemske mastocitoze, a može se razmotriti i kod bolesnika s kožnim i indolentnim oblikom, ako se navedenim lijekovima ne postiže zadovoljavajuća kontrola bolesti. Kao lijekovi izbora za provođenje citoredukcije kod ovih se bolesnika koriste interferon-α2b te nukleozidni analog klorodeoksiadenozin. Monocitna leukemija liječi se po istom principu kao i akutna mijeloidna leukemija. Imatinib mesilat, inhibitor tirozin kinazne aktivnosti, nije se pokazao kao pretjerano učinkovit u liječenju bolesnika s prisutnim c-KIT mutacijama.
Prognoza ovih bolesnika ponajprije ovisi o vrsti mastocitoze. Kožna i indolentna sistemska mastocitoza imaju dobru prognozu te se kod gotovo svih bolesnika uspješno postiže dugotrajna kontrola simptoma. Agresivna mastocitoza i monocitna leukemija imaju lošu prognozu s očekivanim preživljenjem između jedne i tri godine. Sistemska mastocitoza udružena s klonalnom hematološkom bolešću nemastocistne loze ima prognozu koja je uvjetovana hematološkom bolešću.
Autoupalne bolesti predstavljaju široki spektar bolesti koje karakterizira ponavljajuća ili perzistentna sterilna (abakterijska) upala koja nastaje kao posljedica poremećaja ili disregulacije mehanizama nespecifične imunosti uz odsutnost znakova aktivacije mehanizama specifične imunosti (stvaranje specifičnih antitijela, senzibilizacija T-limfocita). Prema načinu pojavljivanja, autoupalne bolesti svrstavamo u dvije skupine: sporadične i nasljedne. Sporadične autoupalne bolesti nastaju kao rezultat kompleksnih interakcija između genetskih promjena i vanjskih čimbenika, dok nasljedni oblici autoupalnih bolesti slijede Mendelove zakone nasljeđivanja (autosomno-dominantno, autosomno-recesivno, X-vezano nasljeđivanje).
Većina ovih bolesti prezentira se već u dječjoj dobi, tako da je prepoznavanje ove skupine bolesti prvenstveno predmet interesa pedijatara. Samo postavljanje dijagnoze ovih bolesti je otežano, što zbog njihovoga rijetkog pojavljivanja, a što zbog širokoga spektra kliničkih manifestacija. Kliničke manifestacije koje mogu upućivati na postojanje autoupalnih bolesti su: promjene na koži (urtikarija bez svrbeža, livedo reticularis, erizipelu slični osipi, granulomi), upale sluznica (afte, ulkusi), promjene na očima (poremećaji vida, periorbitalni edem), serozitisi (perikarditis, pleuritis, peritonitis), artritisi i artalgije te povišeni upalni parametri u fazi kliničkih pogoršanja.
Najpoznatija bolest iz ove skupine je obiteljska sredozemna (ili mediteranska) vrućica.
OBITELJSKA MEDITERANSKA VRUĆICA
Definicija i epidemiologija. Obiteljska mediteranska vrućica (engl. Familial Mediterranean fever, FMF) predstavlja najčešću nasljednu autoupalnu bolest uslijed genskoga poremećaja koji rezultira pojačanim stvaranjem proupalnoga interleukina IL-1β i dovodi do sistemskoga upalnog odgovora koji se u prvom redu očituje rekurentnom vrućicom, kožnim osipom, artritisom i artralgijama te zahvaćanjem seroznih membrana (pleuritis, perikarditis). Bolest je ime dobila zbog povećane incidencije među osobama koje žive u području zemalja Sredozemnoga mora, osobito među Armencima, Arapima, Turcima, Grcima i Talijanima. Prvi simptomi bolesti javljaju se u školskoj dobi te se nastavljaju u odraslu dob, a kod manje od 10 % bolesnika prve manifestacije javljaju se tek u odrasloj dobi.
Etiopatogeneza. FMF nastaje kao posljedica mutacije MEFV gena koji se nalazi na kratkom kraku 16. kromosoma. MEFV gen je odgovoran za stvaranje pirina, proteina eksprimiranoga u granulocitima, monocitima i dentritičkim stanicama, kao i u stanicama seroznih membrana i dermalnim fibroblastima. Mutacije ovoga gena rezultiraju pojačanom aktivnošću enzima kaspaze 1 i pojačanim stvaranjem IL-1β i drugih proupalnih citokina, posljedično čemu dolazi do nastanka upalne reakcije i bez prisutnosti infektivnoga čimbenika. Mutacija MEFV gena prenosi se autosomno-recesivnim putem što znači da je za obolijevanje potrebno da osoba naslijedi mutirani gen od oba roditelja.
Klinička slika. Bolest se dominantno očituje atakama vrućice i drugim kliničkim manifestacijama koje obično spontano prolaze kroz nekoliko dana. Uz povišenu tjelesnu temperaturu bolesnici obično razvijaju i kliničke manifestacije zahvaćenosti kože (erizipelu sličan osip) te seroznih membrana (peritonitis, perikarditis, pleuritis, artritis). Učestalost febrilnih ataka je varijabilna (od jednom tjedno do jednom godišnje ili rjeđe), a nekada može biti potaknuta stresom, infekcijama, cijepljenjem ili, kod žena, menstruacijom. Između ataka se bolesnici osjećaju potpuno zdravo. Kod više od 50 % bolesnika kroz nekoliko se godina razvija reaktivna (AA) amiloidoza koja predstavlja najozbiljniju i najtežu komplikaciju bolesti.
Dijagnoza se postavlja na temelju kliničke slike i anamnestičkih podataka, laboratorijskih nalaza te dokaza mutacije MEFV gena. Tijekom napada bolesti u krvi nalazimo ubrzanu sedimentaciju eritrocita, leukocitozu te povišen C-reaktivni protein. Na FMF treba posumnjati kod svih bolesnika koji su imali najmanje tri epizode kratkotrajne vrućice koje su bile udružene sa serozitisom. Iako je dijagnoza FMF-a klinička, ona se može definitivno potvrditi ispitivanjem mutacije MEFV gena.
Liječenje. Osnovnu terapiju FMF-a predstavlja primjena kolhicina koji može spriječiti opetovane napadaje bolesti i prevenirati razvoj reaktivne amiloidoze. Kolhicin smanjuje adheziju leukocita te djeluje inhibitorno na proizvodnju proupalnih citokina. Kod više od 90 % bolesnika se uz standardnu dnevnu dozu (1,2 - 1,8 mg) postiže kontrola bolesti vrlo brzo nakon uvođenja terapije. Kod bolesnika koji ne reagiraju na kolhicin u obzir dolazi liječenje inhibitorima IL-1 (anakinra, rilonacept, canakinumab).
Definicija. Hemofagocitni sindrom (HFS) predstavlja rijedak, po život opasan i često neprepoznat klinički sindrom kojega karakterizira prejaka aktivacija imunološkoga sustava s posljedičnim oštećenjem različitih tkiva i organa.
Epidemiologija. Prava učestalost ovoga sindroma nije u potpunosti poznata i široko varira u odnosu na zemljopisna područja, tako se HFS češće javlja u područjima s većom učestalošću kosangviniteta, kao npr. u Turskoj gdje ona iznosi oko 7,5:100 000, dok u Švedskoj incidencija iznosi oko 1,2:100 000. Nije utvrđena rasna ili spolna predispozicija za razvoj HFS-a.
Etiopatogeneza. Osnovni patofiziološki poremećaj u HFS-u je nemogućnost eliminacije makrofaga aktiviranih antigenom, zbog čega dolazi do hiperstimulacije imunološkoga sustava. U djetinjstvu su najčešći okidači pojave HFS-a infektivni uzročnici, dok su u odrasloj dobi to i zloćudne te autoimune bolesti. Posljedična prekomjerna aktivacija imunološkoga sustava, osobito makrofaga, NK-stanica i citotoksičnih limfocita, odgovorna je za povećanu produkciju proupalnih citokina koji dovode do tzv. stanja citokinske oluje i višeorganskoga zatajenja. S obzirom na nastanak ovoga sindroma, razlikujemo genetski (primarni) i stečeni (sekundarni) HFS.
Genetski HFS javlja se u dva oblika. Obiteljski oblik nastaje kao posljedica mutacije FLH gena (PRF1, UNC13D, STX11, STXBP2, FLH1) koji su odgovorni za sintezu različitih proteina vezanih uz proces egzocitoze citotoksičnih granula iz upalnih stanica. Također, genetski oblik može biti povezan i s nekim od sindroma imunodeficijencije (rjeđe). Ovaj oblik HFS-a javlja se rano u djetinjstvu (obično potaknut infekcijom) te ima teži i progresivniji tijek s lošijom prognozom.
Stečeni HFS javlja se u starijoj dobi, a osim infekcijama (EBV, CMV, HSV, tuberkuloza), može biti provociran autoimunim (SLE, RA, SS) i zloćudnim bolestima (limfomi i leukemije, karcinom pluća i prostate). Mehanizam nastanka nije u potpuno jasan (poremećaji unutarstaničnih signalnih puteva koji sudjeluju u citotoksičnom odgovoru).
Klinička slika. Bolest karakterizira nagao nastup i često brza progresija, a zbog nespecifičnih manifestacija i sličnosti s drugim bolestima, osobito infekcijama, ostaje neprepoznata. Najčešće kliničke manifestacije HFS-a su vrućica i splenomegalija, ali i hepatomegalija, limfadenopatija, žutica se osip mogu često vidjeti u ovih bolesnika. Kod oko trećine bolesnika prisutne su i neurološke manifestacije koje imaju širok raspon očitovanja (kognitivne smetnje, žarišni ispadi, epilepsija, poremećaj stanja svijesti).
Dijagnoza se postavlja na temelju kliničkih i laboratorijskih kriterija koji uključuju sljedeće kriterije: 1) vrućica, 2) splenomegalija, 3) citopenija koja zahvaća dvije ili više staničnih linija, 4) hipertrigliceridemija i/ili hipofibrinogenemija, 5) povišene vrijednosti feritina (> 500 µg/L), 6) povišen solubilni CD25 - sCD25 (> 2400 U/ml, ELISA test), 7) smanjena ili odsutna aktivnost NK-stanica (protočna citometrija) te 8) hemofagocitoza u koštanoj srži, CNS-u (likvor) ili limfnim čvorovima. Za potvrdu dijagnoze potrebno je imati pet od osam navedenih kriterija. Po postavljenoj dijagnozi potrebno je provesti obradu u smislu dokazivanja uzroka HFS-a (infekcije, maligna bolest, autoimuna bolest).
Liječenje. S obzirom na to da se radi o životno-ugrožavajućem stanju s brzom progresijom, liječenje se provodi u jedinicama intenzivnoga liječenja, a mora se započeti već kod postavljene sumnje na HFS. Liječenje može biti etiološko, simptomatsko i imunomodulatorno. S obzirom na to da se u početku ne može jasno diferencirati uzrok HFS-a, potrebno je svim bolesnicima uključiti antiobiotke širokoga spektra, antivirotike i antimikotike. Simptomatsko liječenje ovisi o pridruženim manifestacijama, a najčešće uključuje transfuzije krvnih pripravaka. Imunomodulatorno liječenje uključuje primjenu glukokortikoida, etopozida, ciklosoprina, intravenskih imunoglobulina i antitimocitnoga globulina prema različitim protokolima. Kod bolesnika s genetskim oblikom HFS-a indicirana je i transplantacija krvotvornih matičnih stanica.
Prognoza. HFS obilježava nepovoljna prognoza koja vrlo često završava smrtnim ishodom, osobito kod bolesnika kod kojih se dijagnoza ne uspostavi i ne započne s mjerama liječenja.
Definicija. Reumatoidni artritis (RA) predstavlja kroničnu, sistemsku upalnu (autoimunu) bolest koja u prvom redu zahvaća sinovijalne ovojnice dovodeći do njihove proliferacije, pojačanoga stvaranja zglobne tekućine te propadanja zglobne hrskavice i pripadajućega koštanog tkiva, što u konačnici rezultira gubitkom funkcije i deformitetom zgloba. Ipak, osim perzistentnoga sinovitisa kao temeljnoga obilježja, RA, kao i druge autoimune bolesti, može zahvatiti i druga, različita tkiva i organe.
Epidemiologija. RA je široko rasprostranjena bolest s prosječnom prevalencijom u odrasloj populaciji između 0,5 i 1 %. Dva do tri puta češće se javlja kod žena nego kod muškaraca, i to obično tijekom četvrtoga i petoga desetljeća kod žena te šestoga i sedmoga desetljeća života kod muškaraca. Sa starenjem se spolne razlike u pojavljivanju bolesti postupno smanjuju i nestaju.
Etiopatogeneza. Uzrok i mehanizam nastanka RA-a nije u potpunosti jasan, ali se zna da se radi o multifaktorskoj genezi koja obuhvaća genetsku predispoziciju, okolišne čimbenike i poremećaj imunološkoga sustava.
Genetska predispozicija. Na genetsku predispoziciju ukazuje povećana učestalost RA-a u nekim obiteljima, kao i između blizanaca, osobito jednojajčanih. Dokazano je da je prisutnost određenih HLA-antigena, kao što su HLA-DR4, HLA-DR1, HLA-DR6 i HLA-DR10, povezana s većom učestalošću RA-a u odnosu na osobe koje nemaju navedene antigene. Osobito snažna povezanost potvrđena je između RA i HLA-DRB1 alela koji kodiraju HLA-DRβ lanac koji sadržava slijed od pet aminokiselina koji se naziva ‘shared epitope’ (SE). Bolesnici koji su heterozigoti za SE imaju teži i erozivni oblik RA-a, dok homozigoti imaju i veći rizik za razvoj ekstraartikularnih oblika bolesti, uključujući vaskulitis. Antigen HLADRB1*04 (HLA- DR4) pojavljuje se kod 50 do 75 % osoba oboljelih od RA-a, a povezan je s težom kliničkom slikom i lošijom prognozom. Osim povezanosti s navedenim HLA antigenima, veću učestalost pojave RA-a imaju i bolesnici s polimorfizom ne-HLA gena, kao što su PTPN22, PADI4, STAT4, TRAF1 i drugi.
Okolišni čimbenici. Različiti se okolišni čimbenici spominju kao mogući pokretači patološkoga imunološkog odgovora koji može završiti nastankom RA-a kod genetski predisponiranih pojedinaca. Kao najznačajniji okolišni čimbenici navode se pušenje (rizik raste s brojem godina pušačkoga staža, kao i s količinom popušenih cigareta, a taj rizik ostaje povišen i do 20 godina nakon prestanka pušenja), infektivni agensi (Mycoplasma spp, Mycobacterium spp, Escherichia coli, Porphyromonas gingivalis, Streptococcus spp, Epstein-Barrov virus, Parvovirus B19, Rubella virus) te izloženost mineralnim uljima i silicijevom oksidu. Povećan rizik imaju i pretile osobe. Kao glavni zaštitni čimbenik danas se navodi primjena oralne hormonske kontracepcije (estrogenski zaštitni mehanizam), iako primjena nadomjesnoga hormonskog liječenja u postmenopauzi ne dovodi do smanjenja rizika od nastanka RA-a.
Poremećaj imunološkoga sustava. Pretpostavlja se da se u RA istodobno radi i o poremećaju staničnoga i o poremećaju humoralnoga imunološkog odgovora na neki antigen. Poremećaj stanične imunosti ogleda se kroz pojačanu aktivaciju T-stanica (osobito Th1 i Th17) koje prevladavaju u sinovijalnoj membrani, a vjerojatno ih aktivira neki vanjski antigen ili promijenjen antigen same sinovijalne membrane (autoantigen). Zbog toga dolazi do pojačane proizvodnje proupalnih citokina među kojima najznačajniju ulogu imaju IL-6, IL1, IL-17 i TNF-α. Oni podržavaju samu upalu dovodeći do kemotaksije upalnih stanica i njihove aktivacije, potiču lučenje proteolitičkih enzima i sl., a sve to rezultira oštećenjem zglobnih (i izvanzglobnih) struktura. Na utjecaj humoralne imunosti ovih bolesnika ukazuje visok postotak bolesnika koji imaju pozitivna specifična autoantitijela, kao što su reumatoidni faktor (IgM antitijela usmjerena na Fc fragment vlastitoga imunoglobulina G) te anticitrulinska antitijela (antitijela usmjerena na ciklički citrulinski peptid). Navedena antitijela imaju sposobnost aktivacije sustava komplementa što u konačnici pridonosi tkivnom oštećenju, pa se i njihova prisutnost povezuje s klinički težim oblicima RA-a i težim oštećenjima zglobne hrskavice i pripadajućega koštanog tkiva.
Patofiziologija. Jednom aktiviran imunološki sustav od strane za sada nepoznatoga antigena (vanjski antigen ili autoantigen) kod genetski predisponiranih osoba dovodi do oštećenja primarno sinovijalne membrane (perzistentni sinovitis) i zglobnih struktura, ali može dovesti i do oštećenja drugih tkiva i organa uslijed oslobađanja proupalnih citokina u cirkulaciju ili taloženjem stvorenih imunokompleksa. Iako glavnu ulogu u cijelom procesu imaju upalne stanice (limfcoiti, neutrofili, monociti, makrofazi), u navedenome oštećenju ulogu imaju i aktivirane endotelne stanice, stanice sinovijalne membrane (hondrociti, fibroblasti) te osteoklasti. Svi oni skupno stvaraju i luče velik broj upalnih medijatora koje ovisno o njihovu učinku dijelimo u nekoliko skupina: citokini, kemokini i faktori rasta (pojedini medijatori imaju višestruku ulogu, pa je i ova podjela uvjetna). Oni imaju lokalni i sistemski učinak. Stimulirane upalne i druge stanice ostvaruju lokalni se učinak na temelju poticanja lučenja proteolitičkih enzima (kolagenaze, metaloproteinaze) koji dovode do enzimatskoga oštećenja ekstracelularnoga matriksa čime dolazi do hrskavične i koštane destrukcije. Karakteristično za RA je i stvaranje panusa, granulacijskoga tkiva koje potječe iz sinovijalne membrane, a prekriva zglobnu hrskavicu i sadrži obilje aktiviranih upalnih stanica i proteolitičkih enzima koji vrše daljnje oštećenje zglobne hrskavice i koštanoga tkiva. Nastali proupalni medijatori i imunokompleksi u sinovijalnoj membrani u određenoj se mjeri oslobađaju i u sistemsku cirkulaciju dovodeći do oštećenja udaljenih organa, kao i do pojave općih sistemskih manifestacija kakve nalazimo i u drugim imunološki posredovanim bolestima.
Patologija. Temeljni znak RA-a je perzistentni sinovitis koji dominantno simetrično zahvaća male zglobove šaka i stopala. Sinovijalna membrana je zadebljana, prožeta upalnim infiltratom u kojemu dominiraju limfociti, makrofazi i plazma stanice. S napredovanjem bolesti nastaje panus koji prekriva zglobne strukture te dovodi do nestajanja hrskavice i destrukcije koštanoga tkiva. Opisani proces zahvaća i pridružene zglobne strukture (zglobna čahura, zglobne sveze) koji također bivaju destruirani. U konačnici upalni proces koji zahvaća zglobove dovodi do gubitka njihove funkcije i nastanka trajnih deformiteta. Karakterističan patohistološki nalaz za RA su i reumatoidni čvorići koji se mogu pojaviti u sinovijalnoj membrani, ali i drugim organima i tkivima, posebice u potkožnom tkivu. Radi se o granulomatoznoj promjeni čije središte čini zona fibrinoidne nekroze okružena histiocitima te slojem limfocita i plazma stanica. Osim zglobova, RA može se manifestirati i afekcijom drugih struktura: akutni nekrotizirajući vaskulitis, fibrinozni poliserozitis, intersticijska fibroza pluća, uveitis i keratokonjuktivitis i dr.
Klinička slika. Kliničke manifestacije koje možemo naći kod bolesnika s RA-om možemo podijeliti u tri skupine: 1) sistemske (opće) manifestacije, 2) zglobne manifestacije te 3) izvanzglobne manifestacije. Kod većine bolesnika bolest počinje postupno (80 %), dok kod manjega broja (20 %) ona može početi kao akutni poliartritis koji zahvaća i velike i male zglobove što ne predstavlja klasičnu kliničku sliku i otežava postavljanje dijagnoze.
Sistemske (opće) manifestacije prisutne su kod gotovo svih bolesnika, a očituju se razvojem slabosti i malaksalosti, anoreksijom i gubitkom tjelesne težine, algičnim sindromom i nejasnim febrilnim stanjem.
Zglobne manifestacije čine temelj kliničke slike reumatoidnoga artritisa. U klasičnom tijeku bolesti, RA najprije simetrično zahvaća male zglobove šaka i stopala, i to ponajprije metakarpofalangealne (MCP), proksimalne interfalangealne (PIP) i metatarzofalangealne (MTP) zglobove, dok su distalni interfalangealni (DIP) zglobovi rijetko zahvaćeni. U kasnijem tijeku bolesti mogu biti zahvaćeni i ručni zglobovi, laktovi, ramena, gležnjevi i kukovi, a temporomandibularni, krikoaritenoidni i sternoklavikularni zglobovi izuzetno su rijetko zahvaćeni. RA može rezultirati i afekcijom gornjega dijela vratne kralješnice (C1-C2), ali za razliku od ostalih seronegativnih spondiloartritisa, ne zahvaća ostatak kralješnice.
Zahvaćenost malih zglobova šaka karakteristična je i najčešće prva manifestacija RA-a (Slika 9.3.). Ona se očituje simetričnim zahvaćanjem MCP- i PIP-zglobova, a bolesnici najprije navode bol i zakočenost koja je najjača u jutarnjim satima, traje dulje od sat vremena i smanjuje se postupnim razgibavanjem. U početnoj fazi bolesti prisutni su znaci upale (oteklina, jutarnja zakočenost, bolnost), dok se napredovanjem bolesti javljaju karakteristični deformiteti: ulnarna devijacija (svi prsti osim palca skreću na ulnarnu stranu), 2) oblik „rupice za gumb“ (fleksija PIP-a, hiperekstenzija DIP-a), 3) oblik „labuđeg vrata“ (hiperekstenzija PIP-a, fleksija DIP-a) te 4) deformacija šake u obliku slova „Z“ (prsti zauzimaju položaj prema ulnarno, a šaka prema radijalno). Ostali zglobovi naginju fleksijskoj kontrakturi.
Slika 9.3. Rani sinovitis MCP- i PIP-zglobova (lijevo), uznapredovali i erozivni RA (desno)
Zahvaćenost malih zglobova stopala također se javlja u ranoj fazi bolesti, a najčešće su zahvaćeni MTP-zglobovi koji su kao i mali zglobovi šaka u ranoj fazi bolesti bolni, otečeni i crveni, dok kasnije dovode do deformiteta kao što su 1) everzija stražnjeg dijela stopala (subtalarni zglob), 2) plantarna subluksacija metatarzalnih glavica, 3) hallux valgus te 4) lateralna devijacija i dorzalna subluksacija palca.
Među ostala zglobna očitovanja, koja su rjeđa kod bolesnika, ubrajamo radijalnu devijaciju ručnoga zgloba te zahvaćanje velikih zglobova kao što su koljena, kukovi, laktovi i ramena. Česta je i pojava sinovijalnih cista u području zahvaćenoga zgloba, najčešće koljena (Bakerova cista). One se manifestiraju kao mekane, fluktuirajuće tumefakcije koje mogu vršiti pritisak na okolne neurovaskularne strukture s posljedičnim cirkulacijskim ili neurološkim ispadima. Iako izuzetno rijetka, zahvaćenost kralješnice najčešće se prezentira subluksacijom C1 i C2 kralješka koja obično prolazi bez većih simptoma, iako nekada može rezultirati pritiskom na kralješničnu moždinu i posljedičnim neurološkim ispadima.
Izvanzglobne manifestacije. RA osim zglobova može zahvatiti i različita druga tkiva i organe: koža, pluća, krvne žile, srce i periferne krvne žile, živčani sustav, oči, bubrege te krv i krvotvorne organe. Osim toga, kod više od 20 % bolesnika s RA-om možemo naći reumatoidne čvoriće, osobito kod bolesnika s pozitivnim reumatoidnim faktorom. Najčešće izvanzglobne manifestacije navedene su u Tablici 9.8.
Reumatoidni čvorići predstavljaju najkarakterističnije izvanzglobne manifestacije RA-a. Oni se najčešće nalaze u potkožnom tkivu u području koštanih prominencija (laktovi, koljena) te u području tetivnih ovojnica (Ahilova tetiva). Klinički se prezentiraju kao okrugle ili ovalne bezbolne čvrste strukture koje su pomične u odnosu na podlogu. Rjeđe se reumatoidni čvorići mogu naći i u plućima, bjeloočnicama i drugim tkivima.
Kožne promjene, osim spomenutih reumatoidnih čvorića, obuhvaćaju i širok spektar promjena koje mogu nastati kao posljedica vaskulitisa (purpura, ulceracije), a česti su i palmarni eritem te piodermija gangrenosum.
Promjene na plućima kod bolesnika s RA-om mogu obuhvaćati široku patologiju: bolesti i poremećaji dišnih puteva (obliterativni bronhiolitis, bronhiektazije), bolesti i poremećaji plućnoga intersticija (plućna fibroza, reumatoidni čvorići) te bolesti i poremećaji pleure (pleuritis, pleuralni izljevi, zadebljanje pleure). Osim toga, ovi su bolesnici skloniji i respiratornim infekcijama, posebice oni na imunomodulatornoj terapiji.
Vaskulitis kod bolesnika s RA-om nastaje kao posljedica odlaganja cirkulirajućih imunokompleksa, a kao jedan od značajnijih rizičnih čimbenika za njegovu pojavnost navodi se pušenje. Vaskulitis se najčešće prezentira kožnim manifestacijama (nekroza, ulceracije, purpura), ali često zahvaća i periferne živce (mononeuritis multipleks), dok su ostale krvne žile rjeđe zahvaćene (koronarne arterije, visceralne arterije).
Kardiovaskularne manifestacije RA-a obuhvaćaju ubrzan razvoj ateroskleroze, perikarditis s ili bez perikardijalnoga izljeva, endomiokarditis te Raynaudov sindom. Rijetka je i pojava reumatoidnih čvorića u području srčanih zalistaka koji mogu dovesti do različitih valvularnih grešaka. Bolesnici s RA-om imaju pet puta veći rizik od razvoja infarkta miokarda u odnosu na opću populaciju.
Kod bolesnika s RA-om karakteristično je zahvaćen periferni živčani sustav, dok središnji živčani sustav uglavnom ostaje netaknut. Najčešće promjene uključuju mononeuritis multipleks kao posljedicu vaskulitisa koji zahvaća krvožilni živčani splet (vasa nervorum), a moguće su i kompresivne neuropatije (sindrom karpalnoga ili tarzalnoga kanala udružen s sinovitisom) te kompresija cervikalne kralješnice uslijed subluksacija i luksacija gornjih cervikalnih kralježaka.
Najčešće očne manifestacije RA-a uključuju kseroftalmiju u sklopu sicca sindroma i keratokonjuktivitis, dok skleritis, episkleritis i skleromalaciju nalazimo rijetko, i to u teškim kliničkim oblicima.
Bolesnici s RA-om mogu razviti i anemiju kronične bolesti ili, rjeđe, hemolitičku anemiju te trombocitozu ili trombocitopeniju. Feltyjev sindrom naziv je za splenomegaliju udruženu s neutropenijom ili pancitopenijom kod bolesnika s RA-om, a obično se javlja kod bolesnika koji imaju pozitivan reumatoidni faktor i anticitrulinska antitijela. Osim toga, moguća je i hepatomegalija i limfadenopatija.
Zahvaćenost bubrega i razvoj kronične bolesti bubrega obično je povezan s amiloidozom uzrokovanim odlaganjem serumskoga amiloidnog proteina A koji predstavlja jedan od reaktanata akutne faze i povišen je kod bolesnika s RA-om.
|
Tablica 9.8. Izvanzglobne kliničke manifestacije RA-a
|
|
Koža
|
Supkutani čvorići, purpura i palmarni eritem (vaskulitis), gangrenozna piodermija
|
|
Pluća
|
Pleuralni izljevi, intersticijska bolest pluća, rematoidni čvorići, plućna hemoragija
|
|
Srce
|
Perikarditis, miokarditis, vaskulitis koronarnih arterija, reumatoidni čvorići
|
|
Oči
|
Keratokonjuktivitis, episkleritis, skleritis, skleromalacija, ulcerativna keratoptija
|
|
Živčani sustav
|
Cervikalna mijelopatija, mononeuritis multipleks (vaskulitis), periferna neuropatija
|
|
Krvotvorni sustav
|
Anemija, trombocitoza, limfadenopatija, Feltyjev sindrom
|
|
Bubrezi
|
Amiloidoza, vaskulitis
|
|
Koštani sustav
|
Osteopenija
|
Dijagnoza RA-a postavlja se na temelju kliničke slike i kliničkoga nalaza, laboratorijske i radiološke obrade te, prema potrebi i kliničkim manifestacijama, i drugim, specifičnim dijagnostičkim metodama.
Laboratorijski nalazi. Kod većine bolesnika s RA-om nalazimo anemiju kronične bolesti te povišene upalne markere (sedimentacija eritrocita, C-reaktivni protein) koji dobro koreliraju s aktivnošću bolesti. Specifični laboratorijski nalazi uključuju određivanje reumatoidnoga faktora (RF) i anticitrulinskih antitijela (ACPA). Reumatoidni faktor pozitivan je kod više od 80 % oboljelih ali nije specifičan jer se može naći i uslijed drugih imunološki posredovanih bolesti te kroničnih infekcija (hepatitis C, tuberkuloza), a može se naći i u do 5 % zdrave populacije. Specifična za RA su anticitrulinska antitijela koja su pozitivna kod oko 70 % oboljelih, a često se mogu otkriti i godinama prije nastupa kliničkih manifestacija. Kod oko 15 % bolesnika s RA ne nalazimo niti reumatoidni faktor niti anticitrulinska antitijela te tada govorimo o seronegativnom RA-u. Bolesnici koji imaju pozitivne navedene serološke markere obično imaju teži klinički tijek i izraženije zglobne manifestacije. Kod manjega broja bolesnika (30 %) mogu se naći i druga autoantitijela, kao što su antinuklearna antitijela (ANA) te antineutrofilna citoplazmatska antitijela (ANCA).
Radiološka dijagnostika. U analizi koštanih zglobnih promjena metodu izbora predstavlja klasična radiološka dijagnostika (klasični rendgenogram šaka i stopala). Unutar prvih šest mjeseci od početka simptoma radiološki je nalaz uglavnom uredan, a prve promjene koje se mogu uočiti su otok mekih periartikularnih struktura i pojava jukstaartikularne demineralizacije. Postupno uslijed propadanja zglobne hrskavice dolazi do suženja zglobnoga prostora te se uočavaju koštane erozije. U razvijenoj fazi bolesti jasno su uočljivi ranije opisani deformiteti. Magnetska rezonanca i ultrazvuk imaju prednost nad dijagnostikom mekotkivnih promjena (npr. rano prepoznavanje sinovitisa), ali njihova uloga u ranoj dijagnostici RA-a još nije u potpunosti utvrđena.
Ostale dijagnostičke metode. Analiza sinovijalne tekućine rijetko se izvodi, većinom u diferencijalno-dijagnostičke svrhe. Dobiveni nalaz kod ovih bolesnika upućuje na povećan broj leukocita (5000 do 100000/mm3) s dominacijom polimorfonukleara. Artroskopija se rijetko izvodi u dijagnostici RA-a, većinom kod atipičnih oblika koji zahvaćaju velike zglobove.
Dijagnostički kriteriji. S obzirom na to da ne postoji laboratorijski niti radiološki nalaz specifičan za RA, danas se dijagnoza temelji na ACR/EULAR kriterijima iz 2010. godine (engl. American College of Rheumatology/European League Against Rheumatism). Kriteriji se temelje na bodovanju zahvaćenosti zglobova, seroloških nalaza, reaktanata akutne faze te trajanju simptoma (Tablica 9.9.). Bodovni sustav se primjenjuje kod svih bolesnika koji imaju zahvaćen barem jedan zglob sa sinovitisom koji se ne može objasniti drugom etiologijom. Konačna dijagnoza RA-a postavlja se ako je ukupan zbroj bodova ≥ 6.
|
Tablica 9.9. ACR/EULAR kriteriji za RA
|
|
Zahvaćenost zglobova
|
|
1 veliki zglob
|
0
|
|
2 - 10 velikih zglobova
|
1
|
|
1 - 3 mala zgloba
|
2
|
|
4 - 10 malih zglobova
|
3
|
|
> 10 zglobova (barem jedan je mali)
|
5
|
|
Serologija
|
|
Negativan RF i negativan ACPA
|
0
|
|
Nisko pozitivan RF ili nisko pozitivan ACPA
|
2
|
|
Visoko pozitivan RF ili visoko pozitivan ACPA
|
3
|
|
Reaktanti akutne faze
|
|
Normalan CRP i SE
|
0
|
|
Povišen CRP ili SE
|
1
|
|
Trajanje simptoma
|
|
< 6 tjedana
|
0
|
|
≥ 6 tjedana
|
1
|
Diferencijalna dijagnoza. U svojoj razvijenoj fazi diferencijalna dijagnoza RA-a ne predstavlja veći problem, ali u svojim počecima i ranoj fazi on može nalikovati na različite bolesti i stanja što može produljiti vrijeme do postavljanja dijagnoze i početka liječenja. U svojim ranim fazama RA može nalikovati na postvirusni artritis (rubeola, hepatitis B, hepatitis C), seronegativne spondiloartropatije, polimilagiju reumatiku te akutni nodalni osteoartritis. Sistemski eritemski lupus i bakterijski endokarditis također mogu nalikovati na RA zbog moguće pojave supkutanih čvorića koji nalikuju na reumatoidne.
Liječenje. Dva su osnovna cilja u liječenju bolesnika s RA-om: simptomatsko olakšanje te očuvanje funkcije i sprečavanje nastanka deformiteta i invaliditeta. S liječenjem treba započeti što ranije, osobito s lijekovima koji modificiraju tijek bolesti (DMARD, engl. disease-modifying antirheumatic drugs) kako bi se na vrijeme zaustavilo napredovanje bolesti koje u konačnici može dovesti do ireverzibilnih oštećenja i trajnih deformiteta. Liječenje treba prilagoditi aktivnosti bolesti koja se procjenjuje različitim kliničkim upitnicima od kojih se najčešće primjenjuje DAS28 (engl. Disease Activity Score 28).
Procjena aktivnosti bolesti. DAS28 bodovni sustav predstavlja matematički izračun čija formula obuhvaća podatke o broju bolnih i otečenih zglobova od ukupno 28 zglobova gornjih ekstremiteta i koljena, sedimentaciji eritrocita i općenitoj procjeni zdravlja bolesnika na vizualnoj analognoj skali od 0 do 100. DAS28 veći od 5,1 predstavlja visoku aktivnost bolesti, manji od 3,2 nisku aktivnost bolesti, a manji od 2,6 remisiju.
Simptomatsko liječenje. Za simptomatsko liječenja bolesnika s RA-om koristimo nesteroidne protuupalne lijekove (nesteroidne anireumatike ili NSAR) i koksibe te glukokortikoide, a oni prvenstveno služe za suzbijanje simptoma bolesti, dok se za kontrolu bolesti kao temelj liječenja koriste DMARD lijekovi, uz preporuku što ranijega uvođenja (odmah po postavljanju dijagnoze), i to prema predloženome modelu „liječenjem do cilja“ (engl. treat-to-target – T2T), kako bi se spriječile ireverzibilne posljedice ove bolesti.
NSAR i koksibi predstavljaju osnovne lijekove koji se koriste kod ovih bolesnika, prvenstveno s ciljem ublažavanja bolova povezanih s aktivnošću bolesti (Tablica 9.10.). Učinak im se temelji na neselektivnoj (COX1 i COX2; NSAR) ili selektivnoj (COX2; koksibi) inhibiciji enzima ciklooksigenaze čime se koči stvaranje prostaglandina, prostaciklina i tromboksana te tako imaju analgetična, protuupalna i antipiretična svojstva. Oni se obično koriste zajedno s DMARD lijekovima, a izbor NSAR ili koksiba (ibuprofen, diklofenak, naproksen, celekoksib) temelji se na individualnom odgovoru bolesniku i podnošenju lijeka. Kod bolesnika s probavnim smetnjama, kao i kod svih starijih od 65 godina, uz NSAR treba primijeniti i gastroprotekciju, najčešće inhibitore protonske pumpe. S obzirom na povećan rizik od nastanka kardiovaskularnih incidenata kod bolesnika koji uzimaju koksibe, ako se primjenjuju, trebaju se izbjegavati kod te skupine bolesnika te ih treba primjenjivati u najnižim dozama. Kod bolesnika koji ne reagiraju na NSAR-e ili koksibe opravdana je i dodatna primjena drugih analgetika, kao što su paracetamol ili dihidrokodein.
|
Tablica 9.10. NSAR i koksibi u liječenju RA-a
|
|
Lijek
|
Doza
|
Interval primjene
|
|
Ibuprofen
|
200 - 400 mg
|
3 - 4 x/dan
|
|
Ibuprofen (sporo-otpuštajući)
|
600 - 800 mg
|
2 x/dan
|
|
Diklofenak
|
25 - 50 mg
|
3 x/dan
|
|
Diklofenak (sporo-otpuštajući)
|
75 - 100 mg
|
1 - 2 x/dan
|
|
Naproksen
|
250 mg
|
3 - 4 x/dan
|
|
Naproksen (sporo-otpuštajući)
|
550 mg
|
2 x/dan
|
|
Celekoksib
|
100 - 200 mg
|
1 - 2 x/dan
|
Glukokortikoidi se također mogu koristiti za postizanje simptomatskoga olakšanja, međutim njihova uporaba bi primarno trebala biti imati ulogu premoštenja do postizanja učinka DMARD lijekova. Iako usporavaju nastanak koštano-zglobnih erozija, ne ubrajaju se u skupinu DMARD lijekova. Za simptomatsko olakšanje zglobnih manifestacija primjenjuju se niske doze kao što su 5 - 10 mg prednizona (ili drugoga ekvivalenta) te se doze veće od 10 mg/dan ne preporučuju. Veće doze opravdane su samo kod izraženih izvanzglobnih manifestacija. Primjenu glukokortikoida treba ograničiti na najkraći mogući period, te je preporuka isključiti ih uz postupno sniženje doze u roku od tri mjeseca zbog mogućih nuspojava kao što su osteoporoza, katarakta, oštećenja tolerancija glukoze i hiperglikemija, kožne promjene, dobitak na težini i dr. U iznimnim slučajevima opravdana je i intraartikularna primjena kortikosteroida (npr. triamkinolon 10 - 40 mg) za kontrolu simptoma, no ona ne smije biti češća od 3 do 4 puta godišnje.
Lijekovi koji modificiraju tijek bolesti (DMARD) su lijekovi čija je primjena povezana s usporavanjem i sprečavanjem nastanka ireverzibilnih, trajnih oštećenja zglobova te izbjegavanjem nastanka trajnih deformiteta koji narušavaju funkciju zglobova i kvalitetu života uopće. Ovi se lijekovi mogu podijeliti u tri skupine: konvencionalni sintetski lijekovi (engl. conventional synthetic DMARDs – csDMARDs), ciljani sintetski lijekovi (engl. targeted synthetic DMARDs –tsDMARDs) te biološki lijekovi (engl. biologic DMARDs – bDMARDs) koje možemo podijeliti na originalne (engl. biologic originator DMARDs – boDMARDs) i bioslične lijekove (engl. biosimilar DMARDs – bsDMARDs). Bioslični lijekovi su biološki lijekovi za koje je dokazana sličnost u pogledu kakvoće, biološke aktivnosti, sigurnosti primjene i djelotvornosti s odobrenim izvornim biološkim lijekom. Danas postoje biosimilari gotovo svih anti-TNFα inibitora te rituksimaba. Ranije primjenjivani lijekovi, kao što su soli zlata, azatioprin, ciklofosfamid i ciklosporin, danas se ne primjenjuju kod reumatoidnoga artritisa.
Konvencionalni sintetski DMARD-i su sintetski lijekovi s imunomodulatornim učincima koji uglavnom nisu u potpunosti razjašnjeni, ali dokazano dovode do smanjenja upale kod ovih bolesnika, što se očituje rezolucijom lokalnih zglobnih promjena, padom vrijednosti reaktanata akutne faze te usporavanjem razvoja ireverzibilnih koštano-zglobnih oštećenja. Za njihov potpun učinak potreban je višemjesečni period (2 - 3 mjeseca), a najbolje rezultate daju kada se međusobno kombiniraju. U kliničkoj praksi najčešće se koriste metotreksat, sulfasalazin, leflunomid te hidroksiklorokin.
Metotreksat je analog folne kiseline koji suprimira aktivaciju T-stanica te inhibira neutrofile. Predstavlja inicijalni DMARD i lijek je izbora za bolesnike s RA-om (tzv. zlatni standard). Metotreksat ostaje temelj liječenja u RA-u, ne samo kao učinkoviti csDMARD, već i kao temelj za kombinaciju s drugim lijekovima, bilo s glukokortikoidima ili drugim csDMARD, bMARD ili tsDMARD lijekovima. Doza metotreksata (bilo u oralnom ili subkutanom obliku) bi trebala biti postupno dizana (u roku od 4 do 6 tjedana) do doze oko 0.3 mg/kg. U našoj populaciji ciljna doza bi trebala biti oko 20 do 25 mg 1 x tjedno (npr. kod Azijata je zbog različite farmakogenetike i manje površine tijela doza manja). Najčešće nuspojave vezane su uz probavni sustav (stomatitis, gastritis), dok su supresija koštane srži, pancitopenija i jetrena lezija rjeđe pojave. Ove su nuspojave češće kod bolesnika s od ranije postojećom bubrežnom insuficijencijom, šećernom bolesti, kao i kod alkoholičara i pretilih bolesnika. Tijekom liječenja metotreksatom bolesnicima se u prosjeku svaka tri mjeseca trebaju kontrolirati krvna slika, jetreni i bubrežni parametri te sukladno nalazima smanjivati dozu lijeka. Primjena folata (1 mg/dan) ili leukovorin kalcija (2,5 - 5 mg 24 sata nakon doze metotreksata) smanjuje mogućnost razvoja navedenih nuspojava te je obavezno primjenjivati ih nakon uzimanja metotreksata. Metotreksat ima i teratogena svojstva te ga ne smiju koristiti trudnice, kao ni osobe u prekoncepcijskoj fazi, a povezan je i s povećanim rizikom od nastanka B-staničnih limfoma i intersticijskoga pneumonitisa (akutni, subakutni). Ako kod bolesnika postoje kontraindikacije ili se razviju nuspojave na metotreksat, preporuka je započeti liječenje s drugim csDMARD- sulfasalazinom ili leflunomidom.
Sulfasalazin je također često korišten lijek u RA-u. Mehanizam djelovanja sulfasalazina nije do kraja razjašnjen, ali se smatra da ima inhibitoran učinak na T-stanice, kao i na ciklooksigenazu, čime smanjuje proizvodnju upalnih medijatora. Koristi se u drugoj liniji liječenja RA-a. Početna peroralna doza iznosi 0,5 mg 2 x/dan uz mogućnost povećanja doze ovisno o kliničkom odgovoru za 0,5 mg svaki tjedan do postizanja kliničkoga poboljšanja ili do postizanja ukupne dnevne doze od 3 mg/dan. Kod 10 do 15 % bolesnika mogu se razviti neutropenija i trombocitopenija, a vrlo rijetko i hemolitička anemija kod bolesnika s deficitom enzima glukoza-6-fosfat dehidrogenaze. Također, bolesnicima s preosjetljivošću na acetilsalicilnu kiselinu nije preporučljivo davati sulfasalazin. Praćenje bolesnika sastoji se od redovnih kontrola krvne slike (prva tri mjeseca jednom mjesečno, a potom svaka tri mjeseca).
Leflunomid je inhibitor sinteze pirimidina koji blokira klonalnu ekspanziju T-stanica čime ostvaruje svoj imunomodulirajući i imunosupresijski učinak. U liječenju RA-a koristi se peroralno u udarnoj dozi od 100 mg/dan kroz tri dana, a potom se nastavlja doza održavanja od 10 do 20 mg/dan. Početak djelovanja lijeka vidljiv je kroz 4 do 6 tjedana. Najčešća nuspojava je proljev, a mogu se javiti i osip, alopecija i hepatotoksičnost (učestalost nuspojava može se smanjiti izostankom primjene udarne doze). Također, lijek ima teratogeno djelovanje te se ne preporuča kod žena reproduktivnoj dobi ili se može primjenjivati uz mjere kontracepcije.
Hidroksiklorokin je također lijek iz skupine csDMARD koji učinak ostvaruje djelujući inhibitorno na AP-stanice (engl. antigen presenting cells) čime smanjuje upalnu reakciju. Kao monoterapija, hidroksiklorokin može biti učinkovit samo u blagim slučajevima, a obično se koristi u kombinaciji s nekim drugim DMARD lijekom (metotreksat ili sulfasalazin). Primjenjuje se peroralno u dnevnoj dozi od 200 do 400 mg. Najozbiljnija nuspojava primjene ovoga lijeka je pigmentni retinitis koji može rezultirati slabljenjem ili gubitkom vida, a javlja se kod oko 2 % bolesnika unutar prvih deset godina liječenja s porastom incidencije nakon tog perioda na 20 % nakon 20 godina liječenja. Zbog te nuspojave svi bolesnici trebaju prolaziti oftalmološki pregled jednom godišnje. Ostale nuspojave uključuju neuropatije i miopatije koje prolaze nakon prekida uzimanja lijeka.
Na početku liječenja bolesnici bi trebali imati redovite kontrole svakih 1 do 3 mjeseca do postizanja cilja liječenja. Ako taj ishod nije postignut, potrebno je promijeniti liječenje povišenjem doze csDMARDs-a, odnosno dodatkom glukokortikoida ili drugih csDMARDs-a. Ako šest mjeseci od početka liječenja nije postignut očekivani ishod, potrebno je razmotriti primjenu bioloških ili ciljanih sintetskih lijekova (u slučaju loših prognostičkih čimbenika terapija se može uvesti i ranije).
Primjena bioloških i ciljanih sintetskih lijekova indicirana je kod bolesnika koji imaju visoku aktivnost bolesti (DAS28 ≥ 5,1 ili DAS28 ≥ 3,2 uz četiri otečena i četiri bolna zgloba bilo koje lokalizacije), unatoč liječenju konvencionalnim sintetskim DMARD lijekovima.
Predstavnici ciljanih sintetskih lijekova (tsDMARD) su JAK-inhibitori. To su male sintetske molekule koje inhibiraju djelovanje JAK-intracelularnih enzima koji prenose signale dobivene od citokina nakon vezanja na receptor na površini stanice te provodeći te signale dovode do aktivatora transkripcije (STAT) koji vode proupalni stanični odgovor (tzv. JAK-STAT put). Predstavnici JAK-inhibitora su tofacitinib, baricitinib i upadacitinib. Po svojoj učinkovitosti slični su bDMARD terapiji. Kod bolesnika liječenih s JAK-inhibitorima zabilježena je pojava tromboembolijskih događaja, stoga je potreban velik oprez kod bolesnika koji su izloženi viskom riziku od duboke venske tromboze ili plućne tromboembolije (uključujući stariju životnu dob, pretilost, tromboembolijski incident u anamnezi, veliki kirurški zahvat ili dugotrajnu imobilizaciju). Ostale neželjene pojave koje se povezuju s primjenom ovih lijeka su povećana sklonost infekcijama i razvojima malignih oboljenja, mijelosupresija i hepatotoksičnost.
Prva linija liječenja biološkim DMARD lijekovima uključuje sve TNF-α inhibitore (originalne i bioslične) i interleukin 6 (IL-6) inhibitore, a u određenim slučajevima i rituksimab.
TNF inhibitori predstavljaju najčešće korištene biološke lijekove u liječenju bolesnika s RA-om. Njihov glavni učinak je inhibicija TNF-α, jednoga od najvažniji upalnih medijatora u patogenezi RA-a. Razlikujemo pet lijekova unutar ove skupine: etanercept (postoje biosimilari), infliksimab (postoje biosimilari), adalimumab (postoje biosimilari), golimumab te certolizumab (doziranje i primjena navedeni su Tablici 9.11). Načelno se ovi lijekovi dobro podnose, mogu stvarati kožne iritacije na mjestu primjene, a ostale, rjeđe i ozbiljnije nuspojave su sklonost infekcijama (kod svih bolesnika prije uključivanja lijeka potrebno je isključiti latentnu tuberkulozu i HBV-infekciju), demijelinizacija, autoimuni fenomeni (kao što je lupus-like sindrom) te povećani rizik od razvoja malignoma. Kontraindicirani su kod bolesnika s umjerenim ili teškim zatajenjem srca (NYHA III i IV). Preoručeno ih je davati u kombinaciji s metotreksatom zbog imunogeničnosti, odnosno moguće pojave stvaranja neutralizirajućih antitijela na lijek, što dovodi do sekundarne neučinkovitosti.
|
Tablica 9.11. Doziranje TNF-inhibitora u liječenju RA-a
|
|
Etanercept
|
50 mg supkutano 1 x tjedno
|
|
Infliksimab
|
3 - 10 mg/kg u obliku iv infuzije koje se ponavljaju nakon 2., 6., 10. i 14., a zatim svakih 8 tjedana
|
|
Adalimumab
|
40 mg supkutano svaki drugi tjedan
|
|
Golimumab
|
50 mg supkutano jednom mjesečno
|
|
Certolizumab
|
200 do 400 mg supkutano svaka 2 do 4 tjedna
|
Interleukin-6 inhibitori su lijekovi koji blokiraju djelovanje interleukina-6 (IL-6), ključnoga proupalnog citokina kod bolesnika s RA-om. Predstavnici ove skupine su tocilizumab (TCZ) i sarilumab (SAR) – inhibirajuća monoklonska antitijela na receptor za IL-6. Njihova prednost je, s obzirom na nisku imunogeničnost, što se mogu davati u monoterapiji, odnosno nije nužna istodobna primjena metotreksata da bi se očuvala učinkovitost. Ovi su lijekovi vrlo učinkoviti u riješavanju sistemskih upalnih simptoma kao što su anemija i umor. Opisane nuspojave su sklonost infekcijama (prije početka liječenja potrebno je isključiti latentnu tuberkulozu i HBV infekciju), divertikulitis, hepatotoksičnost, hematološke poremećaje (neutropenija, trombocitopenija) te hiperlipidemiju.
Ostali biološki lijekovi u liječenju RA-a uključuju abatacept (rekombinantni fuzijski protein koji se veže na kostimulirajuću molekulu CD80 te onemogućava potpuno aktiviranje citoksičnih T-limfocita) i rituksimab (monoklonalno antitijelo usmjereno na CD20 koje dovodi do deplecije B-limfocita). Ovi se lijekovi većinom uključuju kod bolesnika koji ne reagiraju na ranije navedene lijekove. Rituksimab se može dati kao prva linija liječenja ako su prisutne posebne okolnosti, primjerice, kod bolesnika liječenih od limfoma ili kod kojih u anamnezi postoji demijelinizacijska bolest, kod bolesnika s latentnim TBC-om kod kojih je kontraindicirana ili onemogućena primjena kemoprofilakse, kod bolesnika koji dolaze iz područja u kojima je TBC endemijski prisutan te kod bolesnika koji su u anamnezi imali malignu bolest.
Ostali oblici liječenja. Osim lijekova koji čine središnje mjesto u liječenju bolesnika s RA-om, važnu ulogu ima i edukacija bolesnika, provođenje mjera fizikalne terapije te kirurško liječenje (artroplastika, artroskopija) koje je rezervirano za najteže oblike bolesti kod kojih je došlo do razvoja deformiteta i kontraktura.
Prognoza. Ukupno gledano, bolesnici koji se na vrijeme dijagnosticiraju i pravilno liječe, imaju dobru prognozu i ne razvijaju klasične deformitete koji dovode do narušavanja kvalitete života i razvoja invalidnosti. Čimbenici koji predstavljaju prediktore lošijega kliničkog ishoda su: starija životna dob, ženski spol, simetrično zahvaćanje malih zglobova, jutarnja ukočenost zgloba koja traje dulje od 30 minuta, zahvaćenost više od četiri zgloba, pušenje, povišene vrijednosti upalnih parametara te pozitivni reumatoidni faktor i anticitrulinska antitijela.
STILLOVA BOLEST U ADULTNOJ DOBI
Sillova bolest predstavlja sistemski oblik juvenilnoga kroničnog artritisa koji se u malom broju slučajeva može javiti i u odrasloj dobi, najčešće tijekom drugoga i trećeg desetljeća života. Glavna klinička obilježja bolesti su napadaji visoko febrilnoga stanja (> 39 ℃) praćenoga tresavicom i zimicom koji spontano prestaju i bez primjene antipiretika, ružičasti makulopapulozni osip distribuiran po trupu, artritis s artralgijama i mialgijama te generalizirana limfadenopatija. Prava oštećenja zglobova i zglobnih struktura obično se javljaju nekoliko mjeseci po smirivanju općih simptoma. Kod manjega broja bolesnika nađu se i hematosplenomegalija te poliserozitis (pleuritis, perikarditis). U laboratorijskim nalazima obično nalazimo anemiju i izraženu leukocitozu. Dijagnoza se postavlja isključenjem drugih uzroka vrućice, a na bolest treba ukazivati karakterističan osip, uz ostale simptome i znakove. Većina bolesnika dobro reagira na NSAID-e s ili bez kortikosteroida (prednizon). U teškim oblicima koji ne reagiraju na ovo liječenje može se uvesti metotreksat, a u slučaju rezistentnih oblika u obzir dolazi liječenje anakinrom (anti-IL1) ili tocilizumabom (anti-IL6). Najteža i najopasnija komplikacija ove bolesti je sindrom aktivacije makrofaga kojega uz navedene simptome karakterizira i teška pancitopenija s prijetećim komplikacijama.
SISTEMSKI ERITEMSKI LUPUS
Definicija. Sistemski eritemski lupus (SLE) je kronična, multisistemska autoimuna bolest sa širokim spektrom kliničkih očitovanja i mogućnošću zahvaćanja gotovo svih organa, koja može varirati od vrlo blagih do životno-ugrožavajućih oblika, a obilježena je periodima egzacerbacija (tzv. „flair“) i remisija.
Epidemiologija. Procijenjena učestalost SLE-a široko varira te se ona kreće između 20 do 50 oboljelih na 100 000 stanovnika sa značajno češćom pojavnošću kod žena u odnosu na muškarce, kao i kod drugih autoimunih bolesti (odnos žene:muškarci kreće se od 8:1 do 10:1). Učestalost SLE-a veća je i kod osoba crnačke populacije u odnosu na bijelu rasu. Bolest se obično javlja u srednjoj životnoj dobi, najčešće između drugoga i četvrtog desetljeća života.
Etiologija. Etiologija SLE-a uključuje genetske, hormonalne i okolišne čimbenike koji dovode do nepovratnoga sloma imunološke tolerancije vodeći ka stvaranju autoantitijela protiv endogenih nuklearnih i ostalih vlastitih antigena.
Genetska predispozicija. Na ulogu nasljeđa u pojavnosti SLE-a ponajprije ukazuje činjenica da se bolest javlja s povećanom učestalošću unutar pojedinih obitelji te visoka učestalost bolesti kod jednojajčanih blizanaca (i do 60 %). Osim ranije opisivane povećane pojavnosti SLE-a kod osoba koji posjeduju HLA-B8, HLA-DR3 i HLA-DQ antigene, danas je poznat i velik broj drugih gena čije su mutacije i polimorfizmi povezani s većom pojavnošću SLE-a. Radi se o genima koji pripadaju različitim skupinama, a sudjeluju u regulaciji imunoloških reakcija: geni komponenata sustava komplementa (C1q, C2, C4A), geni sustava Fc receptora (FCGR2A, FCGR3A) i TLR receptora (IRF5), geni koji sudjeluju u aktivaciji sustava urođene imunosti (TNFAIP3, ITGAM, IRAK1), geni koji sudjeluju u aktivaciji i signalizaciji limfocita (STAT4, BANK1, BLK, LYN) i dr. Poremećaji navedenih gena rezultiraju povećanom aktivacijom ili oštećenom regulacijom urođenoga ili stečenoga imunološkog odgovora na neki antigen (gubitak tolerancije na vlastite antigene, smanjena eliminacija autoreaktivnih stanica), što je praćeno povećanim stvaranjem interferona tipa I za koji se danas smatra da ima veliku ulogu u samom razvoju bolesti (tzv „interferonski potpis“ ).
Vanjski (okolišni) čimbenici. Do sada su ispitivani brojni okolišni čimbenici koji bi se mogli povezati s razvojem SLE-a, a najuvjerljiviji su virusne infekcije, izloženost ultraljubičastom spektru zračenja te primjena određenih lijekova. U prilog virusnom antigenu kao pokretaču imunološke reakcije kod ovih bolesnika ide činjenica da se kod većine oboljelih mogu naći serološki biljezi preboljele infekcije Epstein-Barrovim virusom te da kod većine bolesnika klinička slika započinje nespecifičnim simptomima nalik prehladi ili gripi. Izloženost ultraljubičastom spektru zračenja (UV-A ili UV-B zrake) dobro je poznat rizični čimbenik za aktiviranje kožnih promjena, a što se tumači oštećenjem DNA epitelnih stanica kože, oksidativnim stresom i apoptozom pri čemu dolazi do povećanoga oslobađanja unutarstaničnih antigena stanica imunološkoga sustava. Neki lijekovi kao što su hidralazin, izoniazid, penicilinamin i prokainamid mogu dovesti do promjena u molekuli DNA s posljedično promijenjenom ekspresijom gena što može dovesti do stvaranja antigena koji mogu potaknuti imunološki proces.
Unutarnji čimbenici. Kao najznačajniji unutarnji čimbenici koji se mogu povezati s nastankom SLE-a spominju se ženski spolni hormoni, posebice estrogen koji, između ostaloga, posjeduje i imunoregulatornu ulogu. U prilog tome govori činjenica da bolest najveću učestalost ima među ženama u reproduktivnoj dobi.
Patogeneza. Pretpostavljeni mehanizam razvoja SLE-a započinje oslobađanjem unutarstaničnih antigena (dijelovi molekule DNA, histoni i sl.) što se događa nakon apoptoze ili oštećenja stanica pri čemu oni bivaju izloženi AP-stanicama (predočne stanice, engl. antigen presenting cells). AP-stanice predočavaju navedene antigene T- i B-limfocitima koji se pri tome aktviraju, a aktivirani B-limfociti stvaraju specifična autoantitijela koja s navedenim antigenima stvaraju imunokomplekse. Oštećenja tkiva i organa nastaju odlaganjem stvorenih imunokompleksa iz cirkulacije ili izravnim vezanjem protutijela za antigene in situ pri čemu dolazi do aktivacije sustava komplementa te kemotaksije i aktivacije neutrofilnih leukocita koji se smatraju temeljnim efektorima oštećenja. Ove su reakcije povezane s povećanom proizvodnjom proupalnih citokina među kojima dominiraju interferon-α i interleukin-10. Sve se više govori i o ulozi drugih stanica u patogenezi SLE-a među kojima se najviše spominju plazmacitne dendritične stanice koje aktiviraju imunokompleksi, a koje potom pojačano stvaraju interferon tip I (interferon-α, interferon-β) i druge proupalne citokine.
Patološka obilježja. SLE može zahvatiti brojna tkiva i organe među kojima su najčešći koža, zglobovi, bubrezi, srce, pluća središnji živčani sustav, serozne membrane (perikard, pleura, peritoneum), probavni sustav i oči. Temeljne histološke značajke zahvaćenih organa su neutrofilna i limfocitna infiltracija uz odlaganje imunokompleksa i nalaz komponenata komplementa, a često se mogu naći i tzv. hematoksilinska tjelešca.
Klinička slika. Kliničke manifestacije SLE-a široko variraju od bolesnika do bolesnika. Većina bolesnika ima izražene opće simptome te znakove zahvaćenosti kože i zglobova, dok su ostale manifestacije nešto rjeđe. Opći simptomi kao što su vrućica nepoznata porijekla, umor, malaksalost, anoreksija te gubitak na težini obično prethode pojavi bolesti, a prisutni su i u vrijeme egzacerbacije bolesti.
Koštano-mišićne manifestacije najčešće su kliničke manifestacije SLE-a i viđaju se kod više od 90 % bolesnika. Najčešće se radi o artralgiji i neerozivnom artritisu koji simetrično zahvaća male zglobove šaka (proksimalni interfalangealni i metakarpofalangealni) koji postaju bolni, ali za razliku od reumatoidnoga artritisa, urednoga su izgleda, iako nekada mogu biti i blaže otečeni i crveni uslijed edema okolnoga mekog tkiva. S obzirom na to da se radi o neerozivnoj upali, nakon smirivanja ne dolazi do zaostajanja deformiteta i trajnih oštećenja i tek kod oko 10 % oboljelih zaostaju deformiteti kao posljedica oštećenja okolnih ligamenata i potpornih struktura (tzv. Jaccoudova artropatija). Kod manjega broja bolesnika moguć je razvoj avaskularne nekroze glave bedrene kosti (što ne mora biti samo posljedica bolesti, nego i produljene primjene kortikosteroida). Mialgija je prisutna kod više od 50 % oboljelih, ali prave znakove miozitisa (laboratorij, histologija) nalazimo kod manje od 5 % bolesnika.
Kožne manifestacije također su česte kod ovih bolesnika te se nalaze u više od 85 % slučajeva (Slika 9.4.). Jedna od najčešćih i najkarakterističnijih kliničkih manifestacija SLE-a je leptirasto crvenilo lica koje podrazumijeva eritematozni makulopapulozni osip distribuiran po periorbitalnom, nosnom i zigomatičnom području, a slične promjene mogu se naći i na drugim dijelovima tijela. Velik dio bolesnika ima i izražen fotodermatitis koji se prezentira pojavom crvenila i bola u područjima kože nakon dužega izlaganja sunčevoj svjetlosti. Rjeđi oblici kožnih manifestacija su livedo reticularis, izolirani osipi na području dlanova i tabana, hiperpigmentacija i alopecija. Kožne promjene obično ne dovode do stvaranja ožiljaka, ali mogu rezultirati atrofijom i hipopigmentacijom. Oblik SLE-a kojega karakteriziraju isključivo kožne promjene bez znakova zahvaćenosti drugih (unutarnjih) organa naziva se diskoidni lupus.
Slika 9.4. Leptirasti eritem (lijevo), livedo reticularis (sredina), diskoidni eritemski lupus (desno)
Visceralne manifestacije. Bubrezi su jedni od najčešće zahvaćenih visceralnih organa u SLE-u (lupusni nefritis), što se može prezentirati širokom kliničkom slikom od asimptomatskih patoloških nalaza urina (proteinurija, hematurija, cilindriurija) do nefrotskoga sindroma i bubrežnoga zatajenja (lupusni nefritis detaljno je opisan u poglavlju Bolesti glomerula). Najčešće promjene na srcu pri SLE-u su perikarditis i endokarditis. Najčešće se radi o simptomatskom ili asimptomatskom perikarditisu koji se očituje izljevom, ali rijetko uzrokuje srčanu tamponadu. Libman-Sacksov endokarditis naziv je za nebakterijski verukozni endokarditis s vegetacijama na mitralnom i trikuspidalnom zalisku, a tipično se javlja kod ovih bolesnika. Bolesnici sa SLE-om imaju i povećan rizik od razvoja ateroskleroze i pratećih kardiovaskularnih komplikacija (akutni infarkt miokarda, cerebrovaskularni incident). Zahvaćenost pluća najčešće se manifestira pleuritisom i pleuralnim efuzijama, dok se pneumonitis i difuzna alveolarna hemoragija značajno rjeđe susreću. Kod oko 30 % bolesnika javljaju se i gastrointestinalne smetenje u vidu mučnine, proljeva i povraćanja, a mogući su i peritonitis, pankreatitis te crijevna pseudoopstrukcija.
Neurološke manifestacije. Zahvaćenost perifernoga i/ili središnjeg živčanog sustava može se naći i kod do 50 % oboljelih od SLE-a, a kliničke manifestacije mogu biti različite. Bolesnici mogu razviti aseptični meningitis, epilepsiju, migrene, cerebelarnu ataksiju, lezije karanijalih živaca ili perifernu senzomotronu polineuropatiju. Kod većine bolesnika izraženi su i psihijatrijski poremećaji u vodu depresije, rjeđe psihoze ili letargije.
Hematološke manifestacije. Uslijed stvaranja autoantitijela na eritrocite, leukocite (posebice neutrofile i limfocite) te trombocite, kod ovih je bolesnika moguće naći i različito izraženu anemiju, leukopeniju te trombocitopeniju. Anemija ne mora biti uzrokovana samo autoantitijelima i hemolizom nego i drugim čimbenicima (anemija kronične bolesti), a prezentira se klasičnim anemijskim sindromom. Leukopenija se očituje kao pojačana sklonost razvoju infekcija, a trombocitopenija krvarenju. Kod trećine bolesnika sa SLE-om nalazimo difuznu perifernu limfadenopatiju.
Očne manifestacije. Zahvaćenost oka najčešće se manifestira kroz retinalni vaskulitis koji uzrokuje brojne infarkte, eksudaciju i krvarenja. Česti su i episkleritis, konjuktivitis i optički neuritis. Potpun gubitak vida nije karakterističan za SLE.
Dijagnoza SLE-a postavlja se na temelju anamnestičkih podataka, kliničke slike i kliničkoga nalaza, općih i specifičnih laboratorijskih nalaza te histološkim pregledom zahvaćenih tkiva ili organa (najčešće koža i bubrezi). Ovisno o indikaciji, odnosno kliničkoj slici, provode se i druge specifične pretrage kojima se dokazuje zahvaćenost pojedinih organa (slikovne radiološke metode, lumbalna punkcija).
Opći laboratorijski nalazi najčešće ukazuju na anemiju tipa anemije kronične bolesti ili hemolize, leukopeniju (posebice neutropeniju i/ili limfopeniju) te umjerenu trombocitopeniju. Sedimentacija eritrocita obično je ubrzana dok je C-reaktivni protein obično uredan (osim ako bolesnik nema i superponiranu infekciju). Ovisno o zahvaćenosti bubrega nalazimo i patološki nalaz urina te različiti stupanj azotemije. Jedan dio bolesnika ima i povišene vrijednosti jetrenih transaminaza.
Specifični laboratorijski nalazi uključuju određivanje specifičnih autoantitijela te vrijednosti komponenata komplementa (C3 i C4). Iako u SLE-u možemo naći širok dijapazon različitih autoantitijela, specifičnima za njega smatraju se antinuklearna antitijela (ANA), anti-dsDNA, anti-Sm, anti-Ro i anti-La (Tablica 9.12). Također kod više od 25 % bolesnika nalaze se i antifosfolipidna antitijela (iako ne moraju svi bolesnici razviti antifosfolipidni sindrom). Određivanje vrijednosti komponenata C3 i C4 korisno je u procjeni aktivnosti bolesti (u aktivnoj bolesti oni su sniženi zbog potrošnje uslijed pojačane aktivnosti sustava komplementa). U tu se svrhu može određivati i ukupna hemolitička aktivnost komplementa (CH50).
|
Tablica 9.12. Autoantitijela u SLE-u
|
|
Antigen
|
Antitijelo
|
Učestalost u SLE
|
|
Komponente jezgre
|
ANA
|
99 %
|
|
dsDNA
|
Anti-dsDNA
|
70 %
|
|
Sm
|
Anti-Sm
|
33 %
|
|
RNP
|
Anti-RNP
|
38 %
|
|
Ro (SSA)
|
Anti-Ro
|
49 %
|
|
La (SSB)
|
Anti-La
|
35 %
|
|
Fosfolipidi
|
Antifosfolipidna antitijela
|
25 %
|
Patohistološka dijagnostika. Biopsija zahvaćenih organa i tkiva ključna je za postavljanje dijagnoze. Obično se u tu svrhu vrši biopsija kožnih promjena ili bubrega. Ključni histološki nalaz podrazumijeva dokaz depozita imunokompleksa i komponenata komplementa metodom imunofluorescencije unutar uzorkovanoga tkiva.
Diferencijalna dijagnoza je široka i uključuje lupus izazvan lijekovima, reumatoidni artritis, sistemski vaskulitis, sistemsku sklerozu, primarni antifosfolipidni sindrom, upalne miopatije, virusni hepatitis, sarkoidozu te akutnu reakciju preosjetljivosti na lijekove.
Dijagnostički kriteriji. U dijagnostici SLE-a danas se koristimo EULAR/ACR klasifikacijskim kriterijima koji su revidirani 2019. godine, a imaju visoku osjetljivost (98 %) i specifičnost (96 %). EULAR/ACR klasifikacijski kriteriji za SLE zahtijevaju pozitivna antinuklearna antitijela (ANA) kao obavezan ulazni kriterij, dok su ostali (dodatni) kriteriji podijeljeni u dvije skupine: klinički (konstitucijski, hematološki, neuropsihijatrijkski, mukokutani, serozni, muskuloskeletni, bubrežni) i imunološki (antifosfolipidna antitijela, proteini komplementa, SLE-specifična antitijela), a boduju se od 2 do 10 bodova (Slika 9.5.). Bolesnici s 10 i više bodova uz ispunjen ulazni kriterij ispunjavaju uvjete za postavljanje dijagnoze SLE-a.
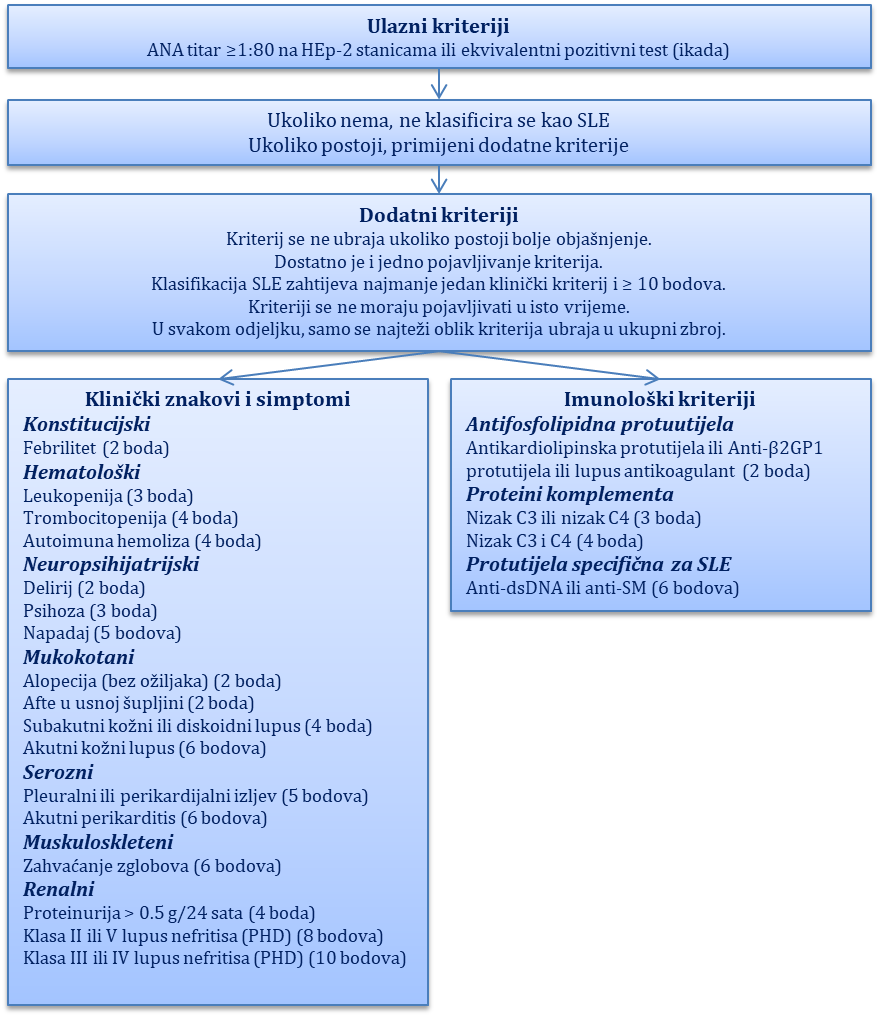
Slika 9.5. EULAR/ACR klasifikacijski kriteriji iz 2019. godine
Liječenje. Terapijski pristup bolesniku sa SLE-om ponajprije ovisi o vrsti i težini kliničkih manifestacija. Svim bolesnicima savjetuje se izbjegavanje potencijalnih uzroka egzacerbacije bolesti što podrazumijeva izbjegavanje prekomjernom izlaganju Suncu i nanošenje zaštitnih krema, izbjegavanje lijekova koji se potencijalno dovode u vezu s pojavom SLE-a i sl. U liječenju SLE-a koriste se antimalarici (hidroksiklorokin), glukokortikoidi, imunosupresivi te biološki lijekovi.
Temelj liječenja koji se preporuča svim bolesnicima sa SLE-om je hidroksiklorokin s ili bez primjene glukokortikoida. Preporučena doza hidroksikolorokina ne bi trebala prelaziti 5 mg/kg tjelesne težine, a zbog moguće retinopatije potrebne su redovite oftalmološke kontrole svakih pet godina. Glukokortikoidi se primjenjuju s ciljem postupne redukcije doze do ≤ 7.5 mg/dan ili njihova ukidanja, s obzirom na njihove brojne nuspojave. Liječenje glukokortikoidima obično započinje intravenskom pulsnom primjenom, najčešće metilprednizolona, a potom slijedi prelazak na peroralni oblik lijeka i brzo smanjivanje doze uz istovremeno rano uvođenje imunosupresivnoga agensa. Visoke doze intravenoznih glukokortikoida (metilprednizolon u dozi od 250 do 1000 mg kroz tri dana) često se koriste u akutnim, organ-ugrožavajućim bolestima (npr. bubrežne, neuropsihijatrijske). Od imunosupresivnih lijekova koriste se metotreksat, azatioprin, mikofenolat-mofetil, ciklofosfamid te kalcineurinski inhibitori, a u zadnjoj liniji liječenja u obzir dolazi i primjena bioloških lijekova (belimumab, rituksimab).
Kao prva linija liječenja kod bolesti kože preporučeni su topički preparati (glukokortikoidi i/ili kalcineurinski inhibitori) te antimalarici, s ili bez sistemskih glukokortikoida. U slučaju nezadovoljavajućega odgovora dodaju se imunosupresivni lijekovi, a u zadnjoj liniji i biološka terapija.
Liječenje neuropsihijatrijskoga oblika SLE-a temelji se na pretpostavljenom patofiziološkom mehanizmu nastanka (upala ili tromboembolijski i ishemijski incident). Kod pretpostavke da se radi o imunološki izazvanom stanju primjenjuju se visoke doze glukokortikoida i imunosupresiva, dok je kod sumnje, odnosno dokazanoga tromboembolijskog incidenta nužna antikoagulantna terapija. Ponekad je potrebno kombinirati obje strategije liječenja. Uz navedene lijekove primjenjuju se i stalni simptomatski lijekovi (antipsihotici, anksiolitici).
Kod hematoloških poremećaja liječenje se sastoji od umjerenih do visokih doza gluukokortikoida u kombinaciji s imunosupresivima (azatioprin, mikofenolat-mofetil, ciklosporin), a kod rezistentnih oblika i intravenozni imunoglobulini i rituksimab.
Kod bolesnika s lupusnim nefritisom uz visoke se doze glukokortikoida u prvoj liniji liječenja za indukciju remisije koriste mikofenolat-mofetil i ciklofosfamid, a u terapiji održavanja remisije mikofenolat-mofetil ili azatioprin. Kod rezistentnih oblika u obzir dolazi i liječenje rituksimabom, a u zadnje vrijeme sve više se govori o uspješnosti liječenja s tzv „multitarget terapijom“, kombinacijom MMF-a i inhibitora kalcineurina.
U najtežih oblika bolesti koji ne ragiraju na gore opisane metode i režime liječenja u obzir dolazi i primjena terapijske plazmafereze (liječenje izbora u trombotičnoj trombocitopeničnoj purpuri, vrlo rijetkoj, ali životno ugrožavajućoj manifestaciji bolesti).
Prognoza bolesnika sa SLE-om uz odgovarajuće liječenje danas je dobra i desetogodišnje preživljenje iznosi preko 90 %. Tijek bolesti često je praćen pojavom egzacerbacija koje zahtijevaju intenziviranje terapije. Bolesnici s afekcijom središnjega živčanoga sustava pokazuju teži tijek bolesti koji slabije odgovara na standardno liječenje. Također, bolesnici sa SLE-om imaju veći rizik od razvoja malignih oboljenja, što je često povezano i sa samom terapijom (imunosupresija), osobito limfoproliferativnih neoplazmi.
Trudnoća i SLE. Bolesnice oboljele od SLE-a obično imaju očuvanu fertilnu sposobnost, osim u teškim kliničkim oblicima, a sam SLE nije kontraindikacija za trudnoću, međutim preporučeno je planiranje trudnoće za vrijeme niske aktivnosti bolesti. Kod bolesnica je nešto češća pojavnost spontanih pobačaja i prijevremenih poroda, osobito ako imaju pozitivna antifosfolipidna antitijela. Trudnoća može dovesti do egzacerbacije bolesti, uz pogoršanje stanja bolesnice. Komplikacije tijekom trudnoće mogu biti maternalne (egzacerbacija lupusa, pogoršanje bubrežne funkcije, pojava ili pogoršanje hipertenzije, razvoj preeklampsije ili venske tromboembolije) te fetalne/novorođenačke (pobačaj, zastoj u intraruterinome rastu, prijevremeni porod, neonatalni lupusni sindrom). Kod trudnica s pozitivnim Anti-Ro i/ili Anti-La antitijelima veća je opasnost od rađanja djece s neonatalnim lupusnim sindromom. Također, kod određenoga broja žena bolest se aktivira šest tjedana nakon poroda, a uzrok ove pojave nije objašnjen. Lijekovi koji se mogu koristiti u trudnoći (u svim tromjesečjima) su hidroksiklorokin, azatioprin, ciklosorin i takrolimus, te glukokortikoidi (preporučljivo u dozi manjoj od 7.5 mg prednizolona dnevno). Također, intravenozni imunoglobulini mogu se koristiti u težim slučajevima. U slučaju pozitivnih antifosfolipidnih protutijela u liječenje se uvode i niskomolekularni heparin i/ili acetilsalicilna kiselina.
Antifosfolipidni sindrom (APS) i SLE. kod bolesnika sa SLE-om češća je pojava antifosfolipidnoga sindroma uzrokovanoga antifosfolipidnim antitijelima (antikardilipinska antitijela - ACA, cirkulirajući lupusni antikoagulant - LAC te anti-β-2-glikoprotein 1 koji se ne određuje rutinski). Opisana antifosfolipidna antitijela usmjerena su na fosfolipidne ostatke na staničnim membranama endotelnih stanica koja ih štite od prekomjernih koagulacijskih poticaja. Potiskivanjem učinka tih fosfolipida, prisutnost antifosfolipidnih protutijela dovodi do prokoagulantnoga stanja zbog čega su ovi bolesnici skloni razvoju arterijskih i/ili venskih tromboembolijskih incidenata, kao i spontanih pobačaja kod trudnica. Dijagnoza se postavlja na osnovi dokazivanja antifosolipidnih antitijela u serumu, a osnovno liječenje sastoji se od tromboprofilakse koju čini primjena niskomolekularnoga heparina ili peroralnih antikoagulantnih lijekova (varfarin, novi antikoagulantni lijekovi nisu preporučeni).
Definicija. Antifosfolipidni sindrom (APS) je autoimuni višesustavni poremećaj karakteriziran arterijskim ili venskim tromboembolijskim događajima u malim krvnim žilama i/ili patologijom trudnoće kao posljedicom prisutnih antifosfolipidnih antitijela (APL), heterogene grupe antitijela usmjerenih protiv proteina koji vežu fosfolipide. On se može javiti kao izolirana bolest (primarni APS) ili u sklopu drugih bolesti (sekundarni APS), najčešće autoimunih (sistemski eritematozni lupus) i malignih (paraneoplastični sindrom). Kod manje od 1 % bolesnika s APS-om može se razviti teška klinička slika praćena višeorganskim zatajenjem - katastrofalni antifosfolipidni sindrom kojega prati visoka stopa smrtnosti.
Epidemiologija. Procjenjuje se da se pojavnost APS-a u općoj populaciji kreće između 2 i 4 % s podjednakom učestalošću primarnoga i sekundarnog oblika. APS se obično javlja u mlađoj populaciji, i to češće kod žena u odnosu na muškarce. Antifofolipidna antitijela mogu biti prisutna i u do 5 % zdrave populacije, a 35 % bolesnika sa SLE-om ima pridružen sekundarni antifosfolipidni sindrom.
Etiopatogeneza. Točan uzrok i mehanizam nastanka bolesti nije u potpunosti jasan, ali se zna da glavnu ulogu imaju antifosfolipidna antitijela. Iako razlikujemo više od dvadesetak različitih vrsta antifofolipidnih antitijela, najznačajnija su lupus antikoagulans (LAC), antikardiolipinska antitijela (ACL) te anti-β2-glikoprotein-I antitijela (aβ2GP-I). Navedena antitijela imaju sposobnost vezanja za fosfolipidne ostatke na membranama endotelnih stanica, trombocita, stranica trofoblasta ili fosfolipida vezanih za faktore koagulacije čime se mijenja njihov fiziološki koagulacijski učinak te dolazi do razvoja koagulacijskih poremećaja, najčešće u vidu prevage učinka prokoagulacijskih mehanizama s posljedičnim trombozama i tromboembolijskim incidentima, a rjeđe može doći do prevage antikoagulacijskih mehanizama s posljedičnim krvarenjima (npr. inhibicija aktivacije protrombina, inhibicija faktora IX). Vezanjem antifosfolipidnih antitijela za endotelne stanice dolazi do njihove aktivacije koja, ne samo da rezultira pojačanom skolnosti razvoju tromboze, nego i kemotaksijom leukocita koji mogu dovesti do pojave endotelne disfunkcije koja ubrzava proces ateroskleroze. “Teorija dva hica” predložena je kao mogući patofiziološki mehanizam, u kojemu sekundarni rizični čimbenici kao što su dob, hipertenzija, dijabetes, pušenje, trudnoća, operacije ili hereditarna trombofilija aktiviraju trombotički učinak antifosfolipidnih antitijela. Najčešće bolesti i stanja u kojima se obično susreću antifosfolipidna antitijela prikazana su u Tablici 9.13.
|
Tablica 9.13. Bolesti i stanja povezana s antifosfolipidnim sindromom
|
|
Autoimune bolesti
|
Hematološke bolesti
|
Primjena lijekova
|
Ostale bolesti/stanja
|
|
Sistemski eritemski lupus
Reumatoidni artritis
Sistemska skleroza
Polimiozitis/dermatomiozitis
Behcetov sindrom
|
Mijeloproliferativne bolesti
Limfoproliferativne bolesti
Paraproteinermije
Hemolitičke anemije
ITP/TTP
|
Prokainamid
Propranolol
Hidralazin
Oralna kontracepcija
Alfa interferon
|
Maligne bolesti
Miastenia gravis
Multipla skleroza
Diabetes melitus
Idiopatske upale crijeva
|
Klinička slika. Najčešća klinička manifestacija APS-a je duboka venska tromboza koja se javlja kod oko 35 % ovih bolesnika, a trećina njih razvije plućnu tromboemboliju. Iako se kod bolesnika s APS-om mogu razviti i venske i arterijske tromboze, venske su nešto češće. Također, česte kliničke manifestacije kod žena s APS-om su komplikacije vezane uz trudnoću, napose opetovani spontani pobačaji te intrauterini zastoj u rastu čeda (uteroplacentarna disfunkcija). Tromboembolijskim incidentima mogu biti zahvaćeni praktički svi organi i tkiva pa se bolest može manifestirati širokim dijapazonom kliničkih sindroma: 1) srce i krvne žile (duboka venska tromboza, akutna okluzija perifernih arterija, akutni koronarni sindrom, intrakardijalna tromboza); 2) pluća (tromboembolija pluća, plućni infarkt), 3) krv i krvotvorni organi (trombocitopenija, autoimuna hemolitička anemija, neutropenija), 4) mozak i periferni živčani sustav (cerebrovaskularni inzult, tromboza kavernoznoga sinusa, transverzalni mijelitis, mononeuritis multipleks), 5) oči (tromboza krvnih žila retine), 6) probavni sustav (tromboza portalne vene, tromboza hepatalnih vena, mezenterijalna ishemija), 7) bubrezi (okluzija renalne vene, tromboza renalnih vena) te 8) komplikacije vezane uz trudnoću (recidivirajući sponatni pobačaji, arterijska hipertenzija uzrokovana trudnoćom, intrauterini zastoj u rastu, HELLP sindrom).
Dijagnoza. Na APS sindrom treba pomišljati kod svih bolesnika koji imaju recidivirajuće tromboembolijske incidente, koji tromboembolijski incident razviju u mlađoj dobi, osobito u odsutnosti provocirajućega čimbenika, kao i kod neuobičajenih lokalizacija i kliničkih prezentacija trombotskih incidenata. Kod svih bolesnika sa sumnjom na APS potrebno je odrediti antifosfolipidna antitijela: lupus antikoagulans (LAC), antikardiolipinska antitijela (ACL) te anti-β2-glikoprotein-I antitijela (aβ2GP-I). U postavljanju dijagnoze APS-a koriste se kriteriji iz 2006. godine (izv. International consensus statement on an update of the clasification criteria for definite antiphospholipid syndrome), utemeljeni na dvije skupine kriterija (klinički i laboratorijski), a bolest je potvrđena ako bolesnik ima pozitivan najmanje jedan klinički i jedan laboratorijski nalaz (prisutnost antitijela u plazmi mora biti dokazana u dva ili više navrata s razmakom od najmanje 12 tjedana) – Tablica 9.14.
|
Tablica 9.14. Dijagnostički kriteriji za antifosfolipidni sindrom
|
|
Klinički kriterij
|
Laboratorijski kriterij
|
|
Vaskularne tromboze koje mogu zahvatiti bilo koje krvne žile (cerebralni vaskularni prsten, koronarne arterije, plućne arterije, arterije ili vene ekstremiteta, hepatalne arterije ili vene, renalne arterije ili vene, retinalne krvne žile i dr.);
Komplikacije u trudnoći: jedan ili više sponatnih pobačaja nakon 10. tjedna gestacije, jedna ili više prijevremeni porod prije ili 34. tjednu gestacije, tri uzastopna neobjašnjena pobačaja prije 10. tjedna gestacije.
|
Pozitivan LAC u 2 različita mjerenja s razmakom od najmanje 12 tjedana;
Pozitivan IgG ili IgM ACL u 2 ili više navrata s razmakom od najmanje 12 tjedana;
Pozitivan IgG ili IgM aβ2GP-I u 2 ili više navrata s razmakom od najmanje 12 tjedana.
|
Liječenje. Klasično liječenje antifosfolipidnoga sindroma uključuje doživotnu primjenu antagonista vitamina K (varfarin) s održavanjem INR-a između vrijednosti 2 i 3 (kod liječenja arterijskih tromboza treba težiti višim vrijednostima). Učinak direktnih antikoagulantnih lijekova kod ovih bolesnika nije ispitana, pa se ni ne primjenjuje u svakodnevnoj praksi. Tijekom trudnoće i za potrebe kirurških zahvata i intervencija bolesnici se prevode na niskomeluklarni heparin. Kod bolesnika s recidivnim tromboembolijskim incidentima unatoč odgovarajućoj antikoagulaciji, kao terapijske opcije u obzir dolaze: povećanje ciljnoga INR na 3 - 4, zamijeniti varfarin niskomolekularnim heparinom, dodati acetilsalicilnu kiselinu, uvesti hidroksiklorokin ili dodati statin.
Katastrofalni antifosfolipidni sindrom predstavlja najtežu kliničku manifestaciju APS-a, a javlja se u manje od 1 % slučajeva. Karakterizira ga prisutnost arterijskih i/ili venskih tromboza u tri ili više različita organa što rezultira višeorganskim zatajenjem. Razvoj ovoga oblika bolesti može biti potaknut infekcijama, kirurškim zahvatima ili primjenom nekih lijekova. Osim nastanka vaskularne tromboze, smatra se da veliku ulogu u kliničkim manifestacijama imaju i povećane količine proupalnih citokina koji se otpuštaju (citokinska oluja). Ovi bolesnici zahtijevaju liječenje u jedinici intenzivnoga liječenja, a ono uključuje intenzivirano antikoagulacijsko liječenje, primjenu glukokortikoida u visokim dozama te plazmaferezu. U liječenju katastrofalnoga APS-a povezanoga s autoimunim oboljenjima u obzir dolazi i primjena ciklofosfamida, intravenskih imunoglobulina, rituksimaba ili ekulizumaba. Unatoč mjerama liječenja, ovo stanje prati visoka stopa smrtnosti (i do 50 %).
SISTEMSKA SKLEROZA (SKLERODERMIJA)
Definicija i epidemiologija. Sistemska skleroza (sklerodermija) predstavlja kroničnu autoimunu bolest vezivnoga tkiva koja zahvaća kožu i unutarnje organe, a obilježena je kroničnom upalom, mikrovaskularnom disfunkcijom i prekomjernim stvaranjem veziva koje dovodi do razvoja progresivne fibroze. Procijenjena učestalost sistemske skleroze u općoj populaciji iznosi 10 do 20 oboljelih na 100 000 stanovnika s većom učestalošću kod žena (odnos žene: muškarci iznosi 4:1). Bolest se obično javlja u dobi između trećega i petoga desetljeća života.
Klasifikacija. Sistemska skleroza javlja se u dva oblika, kao ograničena i difuzna. Ograničena sistemska skleroza primarno zahvaća kožu lica, prstiju i distalnih dijelova ekstremiteta, a naziva se još i CREST sindrom prema kliničkim manifestacijama koje je prate: kalcinoza, Raynaudov fenomen, ezofagealna disfunkcija, sklerodaktilija, teleangiektazije. Difuzna sistemska skleroza primarno zahvaća kožu trupa proksimalne dijelove ekstremiteta (proksimalno od koljena i laktova), a praćena je progresivnijim tijekom i češćim zahvaćanjem unutarnjih organa kao što su pluća, srce i bubrezi, zbog čega je prati teži klinički tijek i lošija prognoza. Ograničena sistemska skleroza nađe se u 70 do 80 % slučajeva, a difuzna sistemska skleroza u 20 do 30 % slučajeva. Sistemsku sklerozu treba razlikovati od lokalizirane sklerodermije koja se odnosi na skupinu benignih kožnih bolesti koje rezultiraju nastankom ograničenih područja skleroze (morfea), uz odsutnost Raynaudova fenomena i zahvaćanja unutarnjih organa. Lokalizirana sklerodermija obično se javlja kod djece.
Etiopatogeneza. Točan uzrok i mehanizam nastanka bolesti nije poznat, ali kao i kod ostalih autoimunih oboljenja, smatra se da ulogu mogu imati genetska predispozicija te utjecaj vanjskih (nepoznatih) čimbenika. Na ulogu genetske predispozicije ukazuje povećana pojavnost bolesti unutar određenih obitelji, kao i povezanost s određenim HLA antigenima (DR1, DR2, DR5, DR52) te promjenama imunoregulatornih gena (STAT4, IRF4, PTPN22, IRAK1, BANK1). Od vanjskih čimbenika sa sistemskom sklerozom najčešće se povezuju virusni antigeni (EBV, CMV), lijekovi (belomicin, pentazocin) te određeni anorganski i organski kemijski pojevi (silicij, polivinilklorid, aromatski ugljikovodici). Predloženi patogenetski mehanizam nastanka sistemske skleroze podrazumijeva oštećenje endotela kod genetski predisponiranih pojedinca kao ključni i ishodišni korak za daljnji razvoj bolesti. Oštećenje endotela rezultira razvojem upalnoga odgovora koji podrazumijeva aktivaciju endotelnih stanica, produkciju proupalnih citokina i kemokina te kemotaksiju i aktivaciju leukocita. Upala rezultira vaskularnim oštećenjem, superponiranom trombozom te tkivnom ishemijom s posljedičnim oslobađanjem unutarstaničnih antigena na koje genetski predisponirani pojedinci mogu stvarati specifična autoantitijela (antinuklearna antitijela, anticentromerna antitijela, antitijela na topoizomerazu I, antitijela na RNA polimerazu III). U ovim prilikama pojačano se stvaraju upalni medijatori (IL-1, IL-4, IL-6, IL-8, TGF-b) koji izravno djeluju na fibroblaste i uzrokuju njihovu aktivaciju. Aktivirani fibroblasti pojačano sintetiziraju kolagen tip I i III, fibronektin i glikozaminoglikane koji se odlažu u okolno tkivo dovodeći do fibroze i strukturalne promjene kože i/ili unutarnjih organa. Prema tome, glavnu patofiziološku osnovu razvoja sistemske skleroze karakterizira trijada koju čine vaskularno oštećenje, poremećen imunološki odgovor te progresivna fibroza.
Patološka obilježja. Početnu fazu sistemske skleroze histološki karakterizira edem i upalni infiltrat koji se pretežno sastoji od limfocita i plazma stanica uz znakove endotelnoga oštećenja, dok u kasnijim fazama nalazimo razvoj fibroze (proliferacija kolagenih vlakana), hijalinizaciju krvnih žila te atrofiju i degeneraciju normalnih staničnih/tkivnih struktura.
Klinička slika. Raynaudov fenomen obično je prva klinička manifestacija obje forme sistemske skleroze i često se pojavljuje i do nekoliko godina prije ostalih kliničkih manifestacija. Opći simptomi kao što su umor, febrilno stanje, anoreksija i gubitak teka jače su i dominantnije izraženi u ograničenom nego u difuznom obliku sistemske skleroze.
Ograničena sistemska skleroza. Kožne promjene kod ovih bolesnika ograničene su na distalne dijelove ekstremiteta, osobito prstiju te lica. Koža na prstima djeluje zategnuto i dovodi do karakterističnih fleksornih deformacija (sklerodaktilija – Slika 9.6.). Koža lica je stanjena i zategnuta, a izgledom dominira ušiljeni nos i mikrostomija (mala usta). Uz te navedene kožne promjene česta je i pojava kalcijskih depozita na vršcima prstiju koji mogu stvarati bolne ulceracije te kožne teleangiektazije. Od unutarnjih organa najčešće su zahvaćeni organi probavne cijevi (najčešće jednjak zbog čega se i javlja poremećaj motiliteta i otežano gutanje) te pluća (kod oko jedne trećine oboljelih od ovoga oblika sistemske skleroze razvija se plućna hipertenzija i/ili intersticijska bolest pluća).
Slika 9.6. Sklerodaktilija
Difuzna sistemska skleroza. Kožne promjene u ovom obliku sistemske skleroze javljaju se u području trupa i proksimalnih dijelova ekstremiteta (proksimalno od koljena, odnosno laktova). Koža najprije postaje edematozna, a kasnije s progresijom postaje atrofična i sklerotična. Klinički je tijek kod takvih bolesnika obilježen pojavom općih simptoma i brzom progresijom te zahvaćanjem različitih unutarnjih organa, najčešće organa probavnoga sustava, pluća, bubrega i srca. Bolesnici sa zahvaćenim probavnim sustavom ponajprije razvijaju smetnje pasaže (pseudoopstrukcija) i malapsorpcijski sindrom. Zahvaćanje bubrega također je česta pojava kod ovih bolesnika (> 50 %) te se manifestira razvojem kroničnoga bubrežnog zatajenja, a moguć je i razvoj renalne sklerodermijske krize koja dovodi do nagloga pogoršanja bubrežne funkcije i razvoja maligne hipertenzivne krize. Zahvaćenost pluća rezultira razvojem plućne fibroze i plućne hipertenzije, a srca restriktivnom kardiomiopatijom, smetnjama provođenja te pojavom konstrikcijskoga perikarditisa.
Sistemska skleroza bez zahvaćanja kože rijedak je oblik sistemske skleroze koja se manifestira zahvaćanjem unutarnjih organa, uz odsutnost kožnih promjena.
Dijagnoza se postavlja na temelju anamnestičkih podataka, kliničke slike i nalaza, laboratorijske i patohistološke dijagnostike te drugih specifičnih dijagnostičkih metoda kojima se utvrđuje zahvaćenost drugih organa i organskih sustava (radiološka dijagnostika pluća, endoskopija probavnoga sustava, MR srca i sl.).
Laboratorijski nalazi. Kod gotovo svih bolesnika može se naći umjerena anemija kronične bolesti, ubrzana sedimentacija eritrocita i povišeni reaktanti akutne faze, osobito kod bolesnika s aktivnom bolesti. Bolesnici sa zahvaćenim bubrezima imaju patološki nalaz urina i povišene vrijednosti metabolita dušika. Antinuklearna antitijela (ANA) pozitivna su kod više od 90 % bolesnika, dok je reumatoidni faktor (RF) pozitivan kod oko 30 % bolesnika. Anticentromerna antitijela (ACA) pozitivna su kod većine bolesnika s ograničenom sistemskom sklerozom, dok su antitijela na topoizomerazu I (anti-Scl-70) i antitijela na RNA polimerazu (anti-RNA-polimeraza) pozitivna kod većine bolesnika s difuznom sistemskom sklerozom. Ostala autoantitijela koja se mogu naći u ovih bolesnika su Anti-Th/To, Anti-PM/Scl, Anti-U1-RNP.
Dijagnostički kriteriji. Kao i kod ostalih bolesti, u postavljanju dijagnoze sistemske skleroze koristimo se različitim dijagnostičkim kriterijima, a najčešće primjenjivani su SSc kriteriji iz 2013. godine koji se temelje na bodovanju osnovnih kliničkih obilježja: zadebljanje kože prstiju obje ruke proksimalno od metakarpofalangealnih zglobova - 9 bodova, zadebljanje kože prstiju distalno od metakrpofalanegalnih zglobova - 4 boda, otečeni prsti - 2 boda, ulkusi na vršcima prstiju - 2 boda, rupičasti ožiljci na vršcima prstiju - 3 boda, teleangiektazije - 2 boda, promjene kapilara ležišta nokta na kapilaroskopiji - 2 boda, plućna hipertenzija - 2 boda, intersticijska bolest luća - 2 boda, Raynaudov fenomen - 3 boda, specifična autoantitijela - 3 boda. Zbroj bodova veći od 9 indikativan je za postavljanje dijagnoze sistemske skleroze.
Diferencijalna dijagnoza. U pogledu diferencijalne dijagnoze kod ovih bolesnika bitno je isključiti druge bolesti i stanja koja se mogu prezentirati Raynaudovim fenomenom, a to su najčešće sistemski eritematozni lupus, miješana bolest vezivnoga tkiva te inflamatorne miopatije. Kožne promjene slične promjenama kod bolesnika sa sistemskom sklerozom mogu imati i bolesnici s eozinofilinim fasciitisom.
Liječenje. Specifičnoga liječenja sistemske skleroze nema i ona se primarno odnosi na simptomatske mjere i ponajprije ovisi o kliničkim manifestacijama bolesti.
U liječenju i kontroli Raynaudovoga fenomena lijekovi izbora su kalcijski blokatori (npr. nifedipin produljenoga djelovanja, 30 - 120 mg/dan), a u slučaju izostanka odgovora u obzir dolazi liječenje sildenafilom (2 x 50 mg/dan) ili u najtežim oblicima intravenskom primjenom iloprosta.
Pasažne smetnje kod ovih bolesnika potrebno je prevenirati uzimanjem manjih, polutekućih ili tekućih obroka uz primjenjivanje ostalih mjera (izbjegavati posjedanje ili lijeganje nakon hranjenja i sl.). U prevenciji refluksa i nastanka refluksnoga ezofagitisa primjenjuju se inhibitori protonske pumpe (npr. omeprazol 20 - 40 mg/dan). Kod razvoja malapsorpcijskoga sindroma uputno je davati antibiotike s ciljem sprečavanja bakterijskoga prerastanja (npr. tetraciklin 4 x 500 mg/dan).
U liječenju hipertenzije i prevencije progresije renalne bolesti preporuča se liječenje inhibitorima angiotenzin-korvetaze (ACE-inhibitori), obično kaptopril u inicijalnoj peroralnoj dozi od 25 mg svakih 6 sati do maksimalno 100 mg svakih 6 sati.
Teži klinički oblici, a osobito ako se radi o zahvaćanju unutarnjih organa, zahtijevaju liječenje glukokortikoidima (prednizon) i/ili imunosupresivima i biološkim lijekovima (ciklofosfamid, metotrekast, mofetil mikofenolat, IL-6 inhibitori). Peroralna primjena prednizona u dozi većoj od 15 mg povezana je s povećanim rizikom od nastanka renalne sklerodermijske krize. U liječenju plućne hipertenzije povezane sa sistemskom sklerozom primjenjuju se i bosentan, riociguat i sildenafil.
Kod bolesnika sa sistemskom sklerozom i fibrozom pluća terapijska opcija su i antifibrotici (nintedanib), koji su prvotno bili odobreni za liječenje idiopatske plućne fibroze.
Prognoza. Bolesnici s ograničenim oblikom sistemske skleroze imaju povoljniju prognozu od onih s difuznim oblikom. Ukupno desetogodišnje preživljenje bolesnika sa sistemskom sklerozom kreće se od 40 do 80 %. Najnepovoljniju prognozu imaju bolesnici s plućnim manifestacijama sistemske skleroze, osobito ako se razvije plućna hipertenzija.
Definicija. Sjögrenov sindrom je sistemska autoimuna bolest koju u prvom redu karakterizira zahvaćenost egzokrinih žlijezda, posebice žlijezda slinovnica i suznih žlijezda, zbog čega su dva temeljna obilježja bolesti suhoća usne šupljine (kserostomija) i suhoća očiju (kseroftalmija), iako ona može zahvatiti i druge organe i tkiva. Sjögrenov sindrom može se javiti kao samostalna i izolirana bolest (primarni Sjögrenov sindrom) ili u sklopu drugih sistemskih autoimunih bolesti kao što su reumatoidni artritis, sistemski eritemski lupus, sistemska skleroza, primarni bilijarni kolangitis i dr. (sekundarni Sjögrenov sindrom).
Epidemiologija. Procijenjena učestalost primarnoga Sjögrenovog sindrom kreće se između 0,1 i 4,8 % unutar opće populacije s izrazitom dominacijom pojavnosti kod žena (odnos žene:muškarci iznosi 9:1). Bolest se većinom pojavljuje između četvrtoga i šestog desetljeća života.
Etiopatogeneza. Uzrok i mehanizam nastanka bolesti do danas nisu u potpunosti razjašnjeni, ali se zna da su povezani s genetskom predispozicijom te poremećenim imunološkim odgovorom u kojemu ulogu imaju stanice urođene imunosti, T-limfociti i B-limfociti.
Na genetsku podlogu bolesti ukazuje veća učestalost kod osoba koje posjeduju HLA-DR3 i HLA-B8 antigene na površini svojih stanica, kao i kod osoba koje imaju mutacije gena koji sudjeluju u proizvodnji interferona za kojega se smatra da ima bitnu ulogu u cijelom patofiziološkom procesu (IRF-5, IL-12A, TNIP1).
Točan pokretač cijeloga procesa do sada nije poznat, ali se smatra da on može biti vanjskoga (virusna infekcija) ili unutarnjeg porijekla (hormonalni disbalans). U prilog virusnoj infekciji govori činjenica da se kod velikoga broja ovih bolesnika u epitelnim stanicama egzokrinih žlijezda mogu naći inkluzije virusnih čestica Epstein-Barrovoga virusa i citomegalovirusa, a u prilog hormonskom utjecaju na razvoj bolesti govori sama činjenica da je bolest daleko češća kod ženskoga spola (utjecaj estrogena kao imunomodulatora).
Predloženi mehanizam razvoja bolesti podrazumijeva aktivaciju stanica urođene imunosti, među kojima se ističu plazmacitne dendritične stanice (PDC) koje počinju u pojačanoj mjeri lučiti interferon-γ, medijator koji izravno djeluje na T- i B-limfocite. Aktivirani T-limfociti infiltriraju ciljne stanice i tkiva (ponajprije egzokrine žlijezde) gdje dovode do izravnih oštećenja dok aktivirani B-limfociti dovode do stvaranja specifičnih autoantitijela. Kod najvećega broja bolesnika nalazimo antinuklearna antitijela na stanične ribosome (anti-Ro ili SS-A, anti-La ili SS-B) te reumatoidni faktor, a rjeđe se mogu naći i anticentromerna antitijela (anti-Ku, anti-p80).
Zahvaćene egzokrine žlijezde bivaju destruirane i zamijenjene vezivnim tkivom što dovodi do gubitka njihove sekretorne funkcije s posljedičnim poremećajima na sluznicama (suhoća sluznica, sklonost infekcijama, ulceracije i sl.). Pored zahvaćanja egzokrinih žlijezda, bolest može zahvatiti i brojne druge organe i tkiva.
Patologija. Kod bolesnika sa Sjögrenovim sindromom dominantno su zahvaćene egzokrine žlijezde među kojima se ističu suzne žlijezde i žlijezde slinovnice, iako mogu biti zahvaćene i druge žlijezde, osobito egzokrine žlijezde probavnoga i dišnog sustava. Glavno histološko obilježje zahvaćenih žlijezda je upalni infiltrat u kojemu dominiraju limfociti i plazma stanice s mjestimičnim stvaranjem germinativnih centara. Žlijezde u konačnici bivaju destruirane, atrofične te prožete vezivnim tkivom. Osim zahvaćanja egzokrinih žlijezda, kod manjega broja bolesnika mogu biti zahvaćena i druga tkiva i organi (koštano-mišićni sustav, koža, pluća, jetra, bubrezi, periferni i središnji živčani sustav).
Klinička slika. Iako su temeljna klinička obilježja Sjögrenova sindroma kseroftalmija i kserostomija, u kliničkoj slici razlikujemo opće simptome te egzokrine i ekstraegzokrine manifestacije.
Opći simptomi nisu posebno izraženi kod ovih bolesnika, a mogu uključivati umor i brzo zamaranje, nepodnošenje napora, gubitak teka i tjelesne težine te nejasna febrilna stanja.
Egzokrine manifestacije temeljna su obilježja ovoga poremećaja. Najčešće zahvaćene egzokrine žlijezde su suzne žlijezde što se manifestira kseroftalmijom te žlijezde slinovnice što se manifestira kserostomijom, a često se zajedno opisuju kao sicca sindrom. Kseroftalmija je naziv za smanjeno stvaranje suza što rezultira suhoćom očne sluznice, osjećajem pijeska i stranoga tijela u oku, a praćeno je učestalim infekcijama, oštećenjima rožnice i fotofobijom (keratokonjuktivitis sicca). Kserostomija je naziv za smanjeno stvaranje sline i posljedičnu suhoću usne šupljine što dovodi do otežanoga gutanja (osobito suhe hrane), osjećaja pečenja te sklonosti razvoju karijesa i gljivičnih infekcija. Zahvaćenost drugih egzokrinih žlijezda je rjeđa, a može imati različite kliničke posljedice, kao što su recidivirajući traheobronhitisi i pneumonije, atrofični gastritis s hipoklorhidrijom, učestale infekcije genitourinarnoga sustava, suhoća vagine i dispareunija i sl.
Ekstraegzokrine manifestacije rjeđe se susreću, a ovise o zahvaćenom tkivu ili organu. One nastaju kao posljedica limfocitne infilitracije i direktnoga oštećenja ili uslijed odlaganja imunkompleksa i aktiviranja sustava komplementa. Najčešće ekstraegzokrine manifestacije Sjögrenova sindroma su: 1) muskuloskeletne (poliartralgije, poliartritis, miopatija, polimiozitis), 2) respiratorne (intersticijski pneumonitis i fibroza, kronični bronitis, bronhiektazije, obliterirajući bronhiolitis s organizirajućom pneumonijom, kronična opstruktivna plućna bolest), 3) renalne (renalna tubularna acidoza tip I, tubularni intersticijski nefritis, glomerulonefritis), 4) neurološke (periferna motorona i senzorna neuropatija, fokalne lezije nalik multiploj sklerozi, disfunkcija kralježnične moždine) te 5) kožne (fotosenzitivne reakcije, purpura, Raynaudov fenomen). Također, uslijed stimulacije limfocita kod ovih je bolesnika daleko češća i pojava ne-Hodgkinovih limfoma (NHL) u odnosu na opću populaciju.
Dijagnoza se postavlja na temelju kliničke slike, općih i specifičnih laboratorijskih nalaza, funkcionalnih testova te biopsije zahvaćene žlijezde (tkiva).
Laboratorijski nalazi. Opći laboratorijski nalazi mogu biti u potpunosti uredni, ali mogu pokazati i srednje izraženu anemiju kronične bolesti, ubrzanu sedimentaciju eritrocita, umjerenu leukopeniju te eozinofiliju. Većina bolesnika ima poliklonalnu hipergamaglobulinemiju, pozitivna antinuklearna antitijela (95 %) te reumatoidni faktor (70 %). Specifična antinuklearna antitijela (ANA) koja nalazimo kod ovih bolesnika su anti-Ro (SS-A) i anti-La (SS-B) koja se smatraju specifičnijima za Sjögrenov sindrom.
Funkcionalni testovi provode se u dokazivanju i procjeni težine kseroftalmije i kserostomije. Za procjenu kseroftalmije najčešće se koristi Schirmerov test kod kojega se u područje vanjske trećine donje vjeđe bez anestetika umeće filter papir te se prati njegovo vlaženje kroz 5 minuta (normala je 15 mm, pozitivan test je ≤ 5 mm). Kserostomija se može objektivizirati sijelografijom ili scintigrafijom.
Patohistološka dijagnostika predstavlja temelj dijagnoze Sjögrenovoga sindroma zajedno s nalazom specifičnih antinuklearnih antitijela. Pri uzorkovanju se najčešće uzima tkivo žlijezda slinovnica, a histološki nalaz koji govori u prilog ovoj bolesti opisan je gore u tekstu.
Dijagnostički kriteriji. Temeljni dijagnostički kriteriji za postavljanje dijagnoze Sjögrenova sindroma su: 1) prisutnost okularnih simptoma (> 3 mjeseca), 2) prisutnost oralnih simptoma (> 3 mjeseca), 3) pozitivni funkcionalni testovi za kseroftalmiju, 4) pozitivni funkcionalni testovi za kserostomiju, 5) pozitivan patohistološki nalaz na Sjögrenov sindrom te 6) pozitivna antinuklerana atitijela (SS-A, SS-B). Dijagnoza je vjerojatna ako je ispunjeno više od tri kriterija, a sigurna ako su pozitivna više od četiri kriterija.
Diferencijalna dijagnoza. Sicca sindrom se, osim u Sjögrenovom sindromu, može se naći i u sklopu drugih, različitih bolesti i stanja kao što su starenje, nuspojava lijekova (antikolinergici, antihistaminici, opijati), anksiodepresivni sindrom, šećerna bolest, amiloidoza, sarkoidoza, limfoproliferativna oboljenja, IgG4 posredovan sijaloadenitis, fibromijalgija te stanje iza radioterapije glave i vrata.
Liječenje Sjögrenovoga sindroma uključuje simptomatske mjere, primjenu imunomodulatornih lijekova te biološke terapije.
Simptomatsko liječenje. Nefarmakološka stimulacija žlijezda kod kserostomije poput korištenje žvaka bez šećera, kiselih bombona za stimulaciju lučenja sline te korištenje umjetne sline trebala bi biti prva linija liječenja, dok je kod kseroftalmije prva linija liječenja primjena umjetnih suza i očnih gelova, čiji je glavni sastojak lubrikant s polimerskom bazom ili viskoznim agensom (metilceluloza, hijaluronat). U slučaju da simptomi perzistiraju unatoč nefarmakološkom pristupu, prelazi se na farmakološku terapiju pri čemu se koriste peroralni preparati muskarinskih agonista kao što su pilokarpin-hidroklorid (4 x 5 mg/dan) ili cevimelin-hidroklorid (3 x 30 mg/dan), a u poboljšanju kontrole očnih simptoma korisnim se pokazala i lokalna primjena 0,05 %-tnoga ciklosporina. Uz to, bolesnici se moraju pridržavati i drugih osnovnih mjera, kao što je izbjegavanje izloženosti štetnim čimbenicima, redovna i odgovarajuća oralna higijena i sl.
Imunomodulatori. Do danas nije dokazana učinkovitost niti jednoga imunomodulatornog lijeka u kontroli simptoma i progresije primarnoga Sjögrenovog sindroma, a pri izraženim sistemskim manifestacijama koristimo se načnima liječenja kao i kod drugih autoimunih bolesti vezivnoga tkiva (glukokortikoidi, hidroksiklorokin, metotreksat, azatioprin). Najčešće primjenjivani i ispitivani lijek u ovoj indikaciji je hidroksiklorokin koji se primjenjuje u dnevnoj dozi do 200 mg. U liječenju neuroloških manifestacija i vaskulitisa mogu se primjenjivati i intravenski imunoglobulini.
Biološka terapija. Dosadašnje studije pokazale su kliničko poboljšanje jedino kod primjene rituksimaba (anti-CD20) i belimumaba (humano monoklonalno antitijelo koje inhibira B-stanični aktivatorski faktor- BAFF), dok se primjena TNF inhibitora (infliksimab, etanercept) nije pokazala uspješnom.
Prognoza primarnoga SS-a uglavnom je povoljna, dok prognoza sekundarnoga SS-a ovisi o kliničkom očitovanju i aktivnosti pridružene autoimune bolesti.
POLIMIOZITIS I DERMATOMIOZITIS
Definicija. Polimiozitis (PM) je naziv za autoimunu bolest nepoznate etiologije koja rezultira upalnim i degenerativnim promjenama poprečno-prugaste (skeletne) muskulature, a kada uz to nalazimo i promjene na koži, govorimo o dermatomizitisu (DM). Uz primarnu zahvaćenost kože i mišića, PM i DM mogu rezultirati i zahvaćanjem drugih tkiva i organa kao što su zglobovi, pluća, srce i probavni sustav. Danas se i PM i DM ubrajaju u heterogenu skupinu idiopatskih upalnih miopatija zajedno s miozitisom s inkluzijskim tjelešcima i imunološki posredovanom nekrotizirajućom miopatijom koji će biti opisani zasebno.
Epidemiologija. Radi se o rijetkim poremećajima s procijenjenom godišnjom incidencijom između 2 i 7 novooboljela bolesnika na milijun stanovnika. Iako se i PM i DM mogu javiti u bilo kojoj životnoj dobi, najčešće se javljaju između petoga i šestog desetljeća života, a žene obolijevaju dva puta češće od muškaraca.
Etiopatogeneza. Iako nepoznat, pretpostavljeni mehanizam nastanka PM-a i DM-a uključuje autoimunu reakciju koja se javlja kod genetski predisponiranih osoba kao odgovor na neki do sada nepoznat antigen. Genetsku predispoziciju za razvoj PM-a i DM-a imaju osobe koje posjeduju određene HLA antigene (HLA-DR3, HLA-DR52 i HLA-DR6), a kao najizgledniji pokretači autoimune reakcije spominju se virusni antigeni (rubeola, influenza, Coxsackie), iako to nije sasvim dokazano. Sam mehanizam nastanka oštećenja tkiva razlikuje se kod PM-a i DM-a. Oštećenja mišićnih vlakana kod bolesnika s PM-om nastaje izravnim citotoksičnim učinkom posredovanim T-limfocitima (CD8+), dok oštećenja mišićnih vlakana i promjene u dermisu kod bolesnika s DM-om nastaju kao posljedica humoralne imunosti, odnosno djelovanja specifičnih protutijela na ciljane antigene pri čemu nastaju imunokompleksi koji aktiviraju sustav komplementa koji dovodi do oštećenja.
Patološka obilježja. Iako su skeletno mišićno tkivo i koža osnovna tkiva koja su zahvaćena ovim poremećajem, zahvaćeni mogu biti i drugi organi, osobito organi probavnoga sustava, zglobovi, srce, pluća i bubrezi. Osnovno histološko obilježje je nalaz mononuklearnoga upalnog infiltrata kojega kod PM-a čine citotoksični CD8+ T-limfociti, a kod DM-a B-limfociti i pomonički CD4+ T-limfociti uz znakove mišićne destrukcije u ranoj fazi (nekroza, miofagocitoza), dok kasnije dolazi do razvoja kroničnih degenerativnih promjena u smislu vezivnoga ožiljkavanja (fibroza). Slične promjene možemo naći i na unutarnjim organima i one rezultiraju specifičnim poremećajima.
Klinička slika. PM i DM mogu imati akutni ili kronični početak. Akutni se prezentira naglo nastalim promjenama unutar nekoliko dana s izraženim općim (sistemskim) simptomima, dok se kronični razvija kroz nekoliko mjeseci i godina, a opći simptomi su manje izraženi. PM i DM također su povezani i s povećanom učestalošću drugih autoimunih bolesti (sistemski eritemski lupus, reumatoidni artritis) kao i s povećanim rizikom od nastanka malignih oboljenja (pluća, želudac, jajnici, dojke).
Polimiozitis. Osnovna klinička manifestacija polimiozitisa je progresivna mišićna slabost koja se razvija tijekom nekoliko tjedana do nekoliko mjeseci. Ona najprije pogađa proksimalne mišiće gornjih i donjih ekstremiteta (najprije donjih), a kasnije i faringolaringealne te respiratorne mišiće. Za razliku od mijastenije gravis, PM ne zahvaća distalne mišiće gornjih i donjih udova, mišiće lica te očne mišiće. Bolesnici se najprije žale na simptome i znakove slabosti proksimalnih mišića donjih i gornjih udova (otežano penjanje uz stepnice, otežano sjedanje i izvođenje čučnjeva, otežano podizanje ruku iznad glave i sl.) koji s vremenom postaju atrofični i mogu dovesti do nastanka kontraktura. U razvijenoj fazi bolesti nalazimo i simptome i znakove vezane uz zahvaćanje faringolaringelanih i respiratornih mišića, kao što je disfagija koja se javlja na samom početku akta gutanja te kronična respiratorna insuficijencija koja može rezultirati potrebom za mehaničkom ventilacijom.
Dermatomiozitis. Kod bolesnika s DM-om nalazimo karakterističan heliotropni eritem lica i edem vjeđa koji se opisuju kao fenomen naočala, a u manjem broju slučajeva oni se mogu širiti i na ostatak lica te presternalnu regiju prsnoga koša. Također, karakteristične su i tzv. Gottronove papule, crvenkasto-ljubičaste ljuskaste papule koje se pojavljuju na ekstenzornim stranama metakarpofalangealnih i interfalanegalnih zglobova te laktova i koljena. Ukoliko se radi o pojavi eritema u razini kože na istim mjestima gdje se pojavljuju i opisane papule, govorimo o Gottronovom znaku. U ostala kožna očitovanja ubrajamo i fotosenzitivan eritem izazvan izlaganjem sunčevoj svjetlosti koji se može javiti u području lica i dekoltea (V-znak), zatiljka i ramena (znak marame, engl. shawl sign) te lateralnim stranama natkoljenice (znak futrole za pištolj, engl. holster sign). Na mjestima kožnih promjena kasnije se mogu razviti hipo- ili hiperpigmentacije, teleangiektazije te atrofični ožiljci. Kod oko trećine bolesnika pojavljuju se i brojni kalcifikati u koži, potkožnom tkivu, fascijama i mišićima. Kožne manifestacije kod većine bolesnika prethode kasnijoj pojavi prethodno opisanih znakova polimiozitisa. Ako se bolesnici prezentiraju samo kožnim promjenama DM-a u trajanju od minimalno 6 mjeseci bez kliničkoga ili laboratorijskog dokaza miozitisa, onda govorimo o amiopatskom dermatomiozitisu (CADM) koji često može biti dio paraneoplastičkoga sindroma.
Ostale kliničke manifestacije. Pored navedenih mišićnih i kožnih očitovanja PM-a, i DM se može očitovati i afekcijom drugih organa. Najčešće se radi o afekciji pluća koja se prezentira nekim od oblika intersticijske patologije (nespecifična intersticijska pneumonija, kriptogena organizirajuća pneumonija, bronhiolitis obliterans organizirajuća pneumonija) koja ima promjenjiv klinički tijek (sporo ili brzoprogresivni) i dovodi do razvoja plućne hipertenzije. Ostala klinička očitovanja uključuju neerozivni artritis i artralgije malih zglobova šaka i stopala, kardiomiopatiju i smetnje provođenja pri afekciji miokarda, vaskulitis i Raynaudov fenomen pri afekciji krvnih žila te manifestacije vezane uz gastrointestinalni sustav (disfagija, gastroezofagealni refluks, malapsorpcijski sindrom). Bolesnici s antitijelima pozitivnim na enzim tRNA sintetazu pokazuju veću vjerojatnost razvoja plućnih i kardijalnih simptoma sa znatno jače izraženim simptomima i znakovima mišićne slabosti, osobito respiratornih mišića, zbog čega je povezana s lošijim kliničkim ishodima i često se izdvaja kao poseban klinički entitet, tzv. antisintetaza sindrom.
Dijagnoza se postavlja na osnovi kliničke slike i kliničkoga nalaza, laboratorijske dijagnostike, funkcionalne mišićne dijagnostike (elektromioneurografija, EMNG) te patohistološkoga nalaza kožnoga i/ili mišićnog bioptata.
Laboratorijska dijagnostika. Najznačajniji laboratorijski nalaz uključuje povišene vrijednosti mišićnih enzima kao što su kreatin-kinaza, mioglobin, aldolaza, a često su povišeni i laktat dehidrogenaza te jetrene transaminaze. Sedimentacija eritrocita i upalni parametri mogu biti uredni, a rijetko se nađe i anemija kronične bolesti. Reumatoidni faktor i antinuklearna antitijela mogu biti pozitivna kod manjega broja bolesnika. Specifična autoantitijela koja se mogu naći kod ovih bolesnika, kao i njihove kliničke značajke prikazani su u Tablici 9.15.
|
Tablica 9.15. Specifična autoantitijela u PM/DM-u
|
|
Autoantitijelo
|
Klinički značaj
|
|
Anti-Jo-1
|
PM/DM s češćom pojavnošću afekcije pluća i zglobova
|
|
Anti-Mi-2
|
Karakterističan za DM
|
|
Anti-MDA5
|
DM s češćim razvojem brzoprogresivne bolesti pluća i kožnim ulceracijama
|
|
Anti-155/140
|
PM/DM udružen s malignim oboljenjima (paraneoplastički sindrom)
|
Elektromioneurografija pokazuje patološki nalaz kod svih bolesnika sa zahvaćenim mišićima. Ona omogućuje uvid u skupinu zahvaćenih mišića (PM/DM ima sklonost zahvaćanja poksimalnih mišićnih skupina gornjih i donjih ekstremiteta).
Biopsija i patohistološki nalaz predstavljaju ključni korak u postavljanju dijagnoze PM/DM-a. Glavne histološke značajke PM-a i DM-a i njihovo razlikovanje opisano je ranije u tekstu (patološka obilježja). Zbog neravnomjerno raspoređenih patoloških promjena unutar zahvaćenoga mišića, moguće je dobiti i lažno negativan rezultat stoga je kod suspektne kliničke slike u takvim slučajevima uputno ponoviti biopsiju.
Ostale dijagnostičke metode. U svrhu potvrde zahvaćanja mišića može se koristiti i ultrazvuk kao jeftina i široko dostupna dijagnostička metoda. Kod akutnoga zahvaćanja mišića nalazimo pojačanu perfuziju i voluminozno tkivo mišića, dok je kod kod kroničnoga miozitisa mišićno tkivo hiperehogeno, slabije perfundirano i volumenom smanjeno. Druga vrlo precizna dijagnostička metoda je MR mišića na kojemu možemo naći simetrični mišićni edem, osobito dobro prikazan u T2 mjernoj slici (MR se osobito koristi kod djece s DM-om kao alternativa biopsiji mišića). Ovisno o drugim kliničkim manifestacijama, potrebno je provesti dijagnostički postupak kako bi se dokazala, odnosno potvrdila zahvaćenost drugih organa i tkiva (radiološka snimka pluća, CT visoke rezolucije pluća, MR i/ili scintigrafija miokarda, endoskopske pretrage probavnoga sustava). Također, s obzirom na relativno čestu povezanost s malignim oboljenjima potrebno je provesti i probir na maligne bolesti.
Dijagnostički kriteriji. Za dijagnostiku PM/DM-a koriste se dijagnostički kriteriji ACR/EULAR (izv. American College of Rheumatology/European League Against Rheumatism) koji se temelje na dobi, znacima mišićne slabosti, prisutnosti kožnih i ostalih manifestacija, laboratorijskim pokazateljima te patohistološkim znacima ako je obavljena biopsija mišića (Tablica 9.16.). Definitivna dijagnoza PM/DM-a postavlja se ukoliko je ukupni zboj bodova veći od 7,5 ako nije rađena biopsija, odnosno veći od 8,5 ako je rađena biopsija.
|
Tablica 9.16. ACR/EULAR dijagnostički kriteriji za PM/DM
|
|
Kategorija
|
Bodovi (s biopsijom)
|
Bodovi (bez biopsije)
|
|
Početak simptoma u dobi od 18. do 39. godine života
|
1,3
|
1,5
|
|
Početak simptoma nakon 40. godine života
|
2,1
|
2,2
|
|
Progresivna simetrična slabost proksimalnih mišića ruku
|
0,7
|
0,7
|
|
Progresivna simetrična slabost proksimalnih mišića nogu
|
0,8
|
0,5
|
|
Mišići fleksori vrata slabiji su od ekstenzora
|
1,9
|
1,6
|
|
Proksimalna muskulatura nogu je slabija od distalne
|
0,9
|
1,2
|
|
Heliotropni osip
|
3,1
|
3,2
|
|
Gottronove papule
|
2,1
|
2,7
|
|
Gottronov znak
|
3,3
|
3,7
|
|
Disfagija, poremećaj motorike jednjaka
|
0,7
|
0,6
|
|
Pozitivna Anti-Jo-1 autoantitijela
|
3,9
|
3,8
|
|
Povišene vrijednosti CK ili LDH ili AST ili ALT
|
1,3
|
1,4
|
|
PHD: infiltracija endomizija mononuklearima, bez invazije
|
|
1,7
|
|
PHD: perimizijalna i perivaskularna invazija mononuklearima
|
|
1,2
|
|
PHD: perifascikularna atrofija
|
|
1,9
|
|
PHD: obrubljene vakuole
|
|
3,1
|
Liječenje. Osnovu liječenja PM/DM čine glukokortikoidi, dok u drugoj liniji liječenja koristimo i druge imunosupresive, intravenske imunoglobuline te biološku terapiju. Simptomatsko liječenje uključuje primjenu analgetika i nesteroidnih protuupalnih lijekova, izbjegavanje prekomjernoga izlaganja Suncu i korištenje zaštitnih čimbenika te provođenje mjera fizikalne terapije.
Prva linija liječenja. Glukokortikoidi predstavljaju prvu liniju liječenja bolesnika s PM/DM-om. Ona obično započinje peroralnom primjenom prednizona u dozi od 0,5 - 1 mg/kg/dan kroz mjesec dana kada se postupno, ovisno o kliničkom odgovoru, doza smanjuje do doze potrebne za održavanje remisije (5 - 15 mg/dan ili 10 - 25 mg svaki drugi dan). U teškim kliničkim oblicima opravdana je i primjena pulsne intravenske glukokortikoidne terapije koju čini primjena 500 do 1000 mg metilprednizolona intravenski na što se nastavlja peroralna primjena prednizona. Kod bolesnika koji su na dugotrajnoj terapiji glukokortikoidima potrebno je provoditi mjere prevencije razvoja osteoporoze i infekcija.
Druga linija liječenja. Kod bolesnika koji ne reagiraju na terapiju glukokortikoidima opravdana je primjena drugih imunosupresiva (metotreksat, azatiprin, ciklosoprin, ciklofosfamid, hidroksiklorokin, mikofenolat mofetil, takrolimus) i biološke terapije kao zadnje opcije (TNF inhibitori, rituksimab, anakinra). Kod vitalno ugroženih bolesnika s bolešću rezistentnom na primjenu ovih lijekova mogu se primjenjivati i intravenski imunoglobulini. Primjenjivane doze prikazane su u Tablici 9.17.
|
Tablica 9.17. Druga linija liječenja PM/DM
|
|
Lijek
|
Doza
|
|
Metotreksat
|
do 25 mg/tjedno per os/supkutano
|
|
Azatiorpin
|
1,5 - 2 mg/kg/dan per os
|
|
Ciklosporin
|
2 - 3,5 mg/kg/dan per os
|
|
Mikofenolat mofetil
|
1 - 2 g/dan per os
|
|
Takrolimus
|
2 - 3 mg/dan per os; 0,1 % mast topički
|
|
IVIg
|
2 g/kg intravenski podijeljeno kod više dnevnih doza
|
Prognoza. Većina bolesnika s PM/DM-om imaju dobru prognozu, ali zahtijevaju dugotrajno imunosupresivno liječenje. S obzirom na povećan rizik od razvoja malignih bolesti, ove bolesnike treba češće kontrolirati i provoditi mjere probira na maligna oboljenja.
OSTALE IDIOPATSKE UPALNE MIOPATIJE
Miozitis s inkluzijskim tjelešcima ubraja se u skupinu idiopatskih upalnih miopatija koje se javljaju u starijoj životnoj dobi (nakon petoga desetljeća života) i dominantno zahvaća osobe muškoga spola. Uzrok i mehanizam nastanka bolesti nisu poznati, a vjeruje se da kao i ostale bolesti iz ove skupine nastaje udruženim djelovanjem genetskih i vanjskih čimbenika. Bolest karakterizira sporoprogresivan tijek, a najčešće zahvaća m. quadriceps femoris te fleksore prstiju šaka što rezultira razvojem atrofije specifičnih lokalizacija (medijalna strana natkoljenica, ventralna strana podlaktica). Vrijednosti kreatin kinaze su uredne ili blago povišene, a kod većine bolesnika mogu se naći specifična anti-cN1A antitijela. Definitivna dijagnoza se postavlja biopsijom mišića u kojima nalazimo specifična inkluzijska tjelešca (obrubljene vakuole). Liječenje se temelji na primjeni kortikosteroida, ali je odgovor slab.
Imunološki posredovana nekrotizirajuća miopatija rijedak je oblik idiopatske upalne miopatije koja se javlja u starijoj dobi, a klinički je obilježena slabošću proksimalnih mišića kao i kod PM/DM-a uz karakterističan nalaz multifokalnih nekrotizirajućih žarišta u bioptatu zahvaćenoga mišića. Ovaj oblik upalne miopatije često se javlja kao paraneoplastični sindrom. Kod većine ovih bolesnika nalazimo pozitivna anti-HMGCR i anti-SRP autoantitijela. Klinička slika je nespecifična (slabost proksimalnih skupina mišića donjih i gornjih ekstremiteta), a osnovu dijagnoze čini patohistološki nalaz. Liječenje se provodi kao i kod PM/DM-a.
MIJEŠANA BOLEST VEZIVNOGA TKIVA
Definicija. Miješana bolest vezivnoga tkiva (MCTD, engl. mixed connective tissue disease) predstavlja klinički entitet koji podrazumijeva međusobno preklapanje kriterija više autoimunih bolesti vezivnoga tkiva (sistemski eritemski lupus, sklerodermija, polimiozitis/dermatomiozitis) kod bolesnika koji imaju visok titar antinuklearnih antitijela na ribonukleoproteinski antigen (anti-U1RNP).
Epidemiologija. Prevalencija MCTD-a je različita u različitim krajevima, ali se u prosjeku kreće u okviru od oko 3 oboljela na 100 000 stanovnika. Žene znatno češće obolijevaju u odnosu na muškarce, a vršna incidencija pojavnosti je u drugom i trećem desetljeću života.
Etiopatogeneza. Točan uzrok i mehanizam razvoja bolesti nije poznat. Genetsku predispoziciju čini povezanost s HLA-DR4 i HLA-DQ3 antigenima. Trenutna saznanja govore u prilogu abnormalnom odgovoru T- i B-limfocita (stanično i humoralno posredovani imunoodgovor) na modificirane vlastite antigene koji mogu dovesti do oštećenja različitih tkiva i organa stvarajući široki dijapazon kliničkih manifestacija.
Klinička slika. Kliničke manifestacije MCTD-a su različite i mogu uključivati različite poremećaje i očitovanja: Raynaudov fenomen (96 %), artralgije i artritis (96 %), poremećaj motorike jednjaka (66 %), plućne manifestacije (66 %), otekline ruku (66 %), nespecifičan osip (53 %), miozitis (51 %), sklerodaktilija (49 %), poliserozitis (43 %) te plućna hipertenzija (23 %). Najraniji znak koji godinama prethodi razvoju drugih manifestacija je Raynaudov fenomen i prisutan je kod gotovo svih bolesnika. Tijekom vremena klinička slika može nalikovati više jednoj bolesti te može postupno mijenjati svoj karakter i postajati sličnija drugoj bolesti vezivnoga tkiva.
Dijagnoza se postavlja na temelju kliničke slike i nalaza, laboratorijske i druge specifične obrade. Na MCTD treba pomišljati kod bolesnika koji imaju raznoliku kliničku sliku koja bi istodobno mogla odgovarati većem broju bolesti vezivnoga tkiva te imaju visok titar anti-U1RNP autoantitijela. Ostali laboratorijski nalazi mogu pokazivati poliklonalnu hipergamaglobulinemiju, pozitivna antinuklearna antitijela (ANA), pozitivan reumatoidni faktor (RF) uz različit stupanj anemije kronične bolesti i trombocitopenije. U fazi aktivne bolesti nalazimo i ubrzanu sedimentaciju eritrocita te povišeni C-reaktivni protein.
Liječenje ovih bolesnika slično je liječenju bolesnika sa SLE-om, a temelji se u prvom redu na kontroli simptoma: Raynaudov fenomen (blokatori kalcijskih kanala), artritis i artralgije (nesteroidni protuupalni lijekovi, hidroksiklorokin, niske doze glukokortikoida ili metotreksata), poremećaj motorike jednjaka i gastroezofagealni refluks (inhibitori protonske pumpe), plućna hipertenzija (inhibitori fosfodiesteraze, antagonisti endotelinskih receptora, prostaglandini) i dr.
Prognoza u prvom redu ovisi o težini kliničkih manifestacija. U načelu je desetogodišnje preživljenje veće od 80 %, a plućna hipertenzija predstavlja glavni negativni prognostički znak i glavni uzrok smrtnosti u ovih bolesnika.
Definicija. Vaskulitis je naziv za heterogenu skupinu bolesti i poremećaja koje karakterizira upala stijenke krvnih žila, a mogu se javiti kao izolirana sistemska bolest (primarni vaskulitis) ili u sklopu neke druge bolesti ili poremećaja (sekundarni vaskulitis).
Klasifikacija. Klasično primarne vaskulitise klasificiramo prema veličini krvnih žila koje zahvaćaju, pa tako razlikujemo vaskulitise velikih, srednjih i malih krvnih žila, no današnje spoznaje u klasifikaciju uzimaju i druge bitne karakteristike kao što su: epidemiološke karakteristike bolesnika (spol, dob, etnička pripadnost), sklonost zahvaćanju određenih organa (organotropizam), patohistološki nalaz (prisutnost granuloma, imunokompleksa, pauci-imuni vaskulitis) te prisutnost ili odsutnost antineutrofilinih citoplazmatskih protutijela (ANCA) u serumu bolesnika. S obzirom na navedene karakteristike, možemo razlikovati nekoliko skupina vaskulitisa (Tablica 9.18, prema International Chapel Hill Consensus Conference of the Nomenclature of Vasculitides, 2012.).
|
Tablica 9.18. Klasifikacija vaskulitisa
|
|
Vaskulitisi velikih krvnih žila
Takayasu arteritis
Gigantocelularni vaskulitis
|
|
Vaskulitisi srednje velikih krvnih žila
Nodozni poliarteritis
Kawasakijeva bolest
Buergerova bolest
|
|
Vaskulitisi malih krvih žila povezani s antineutrofilinim citoplazmatskim antitijelima (ANCA pozitivi vaskulitisi)
Mikroskopski poliangiitis
Granulomatoza s poliangiitisom ili Wegenerova granulomatoza
Eozinofilna granulomaoza s poliangiitisom ili Churg-Straussov sindrom
|
|
Vaskulitisi malih krvnih žila posredovani imunompleksima
Bolest glomerularne bazalne membrane ili Goodpasteureov sindrom
IgA vaskulitis ili Henoch-Schonleinova purpura
Krioglobulinemijski vaskulitis
Hipokomplementarni urtikarijski vaskulitis
|
|
Vaskulitisi koji varijabilno zahvaćaju krvne žile
Behcetov sindrom
Coganov sindrom
|
|
Vaskulitisi koji zahvaćaju samo jedan organ
Kožni leukocitoklastični angiitis
Kožni arteritis
Primarni angiitis središnjeg živčanoga sustava
Izolirani aortitis
|
|
Vaskulitisi udruženi sa sistemskim upalnim bolestima
Lupusni vaskulitis
Reumatoidni vaskulitis
Sarkoidni vaskulitis
|
|
Vaskulitisi udurženi s ostalim bolestima i stanjima
Hepatitis B i C, sifilis
Lijekovima izazvan vaskulitis posredovan imunokompleksima ili ANCA antitijelima)
Ostali (paraneoplastični sindrom, subakutni infektivni endokarditis)
|
GIGANTOCELULARNI ARTERITIS (TEMPORALNI ARTERITIS)
Definicija i epidemiologija. Gigantocelularni arteritis predstavlja sistemski panarteritis koji dominanto zahvaća velike i srednje velike krvne žile, a obično je povezan s polimijalgijom reumatikom zbog čega se često opisuju i kao dva spektra iste bolesti. Gigantocelularni arteritis dominantno zahvaća ekstrakranijalne ogranke karotidne arterije među kojima se najviše ističe temporalna arterija. Bolest je dobila ime po karakterističnom patohistološkom nalazu multinuklearnih orijaških (divovskih) stanica u upalno promijenjenim krvnim žilama. Bolest se obično javlja kod osoba starijih od 50 - 60 godina (prosječna dob pri postavljanju dijagnoze iznosi 70 godina) s nešto češćom pojavnošću kod žena u odnosu na muškarce (odnos 3:1).
Etiopatogeneza. Točan uzrok i mehanizam nastanka bolesti do sada nije razjašnjen. Smatra se da upala nastaje kao posljedica prisustva određenoga antigena (Varicella Zoster, Burkholdelia pseudomallei) u adventiciji krvnih žila koji aktivira AP-stanice (koje predočuju antigen) koje potom luče IL-18 te aktiviraju Th1 i Th 17 subpopulaciju limfocita koji potom luče INF-γ i IL-17. Ovaj imunološki odgovor rezultira kemotaksijom i stimulacijom makrofaga koji formiraju multinuklearne (orijaške) stanice. Multinuklearne stanice imaju sposobnost stvaranja i lučenja metaloproteinaza, proupalnih citokina (najistaknutiji je IL-6) te faktora rasta, a posljedično tome dolazi do destrukcije elastične membrane, hiperplazije intime i glatkomišićnih stanica, stvaranja granuloma te zadebljanja krvne žile u cijelosti (transmuralna upala). U području zahvaćene krvne žile može doći do superponirane tromboze i nastanka okluzije s posljedičnom distalnom ishemijom. Povećana povezanost između pojavnosti ovoga vaskulitisa i HLA-DRB1*04 haplotipa ukazuje i na genetsku predispoziciju u njegovom razvoju.
Patologija. Gigantocelularni arteritis najčešće segmentalno zahvaća ekstrakranijalne ogranke karotidne arterije (a. temproralis, , a. veretbralis, a. ophtalmica i a. ciliaris posterior), ali može zahvatiti i njezine intrakranijalne ogranke i aortu i njezine glavne ogranke (rjeđe). Makroskopski zahvaćeni dio arterije je zadebljan, a mikroskopski promjene nalazimo cijelom debljinom stijenke (transmuralna upala), a najznačajnije su formiranje granuloma, prekid elastične membrane i zadebljanje intime koja rezultira suženjem i zatvaranjem lumena. Cijela stijenka prožeta je limfocitima, epiteloidnim i multinuklearnim (orijaškim) stanicama.
Klinička slika. Gigantocelularni arteritis karakteriziraju opći simptomi, simptomi i znakovi zahvaćenih arterija te polimijalgija reumatika koja se javlja kod više od polovine oboljelih. Opći simptomi uključuju vrućicu nejasnoga porijekla, umor i malaksalost, gubitak teka i mršavljenje uz apatičnost i bezvoljnost.
Simptomi i znakovi arteritisa. S obzirom na to da su najčešće zahvaćeni ekstrakranijalni ogranci karotidne arterije, bolest se najčešće prezentira jakom glavoboljom, osjetljivošću vlasišta i kože lica, poremećajima vida (diplopije, gubitak vida) te bolovima u donjoj čeljusti, osobito pri žvakanju. U rjeđim oblicima može biti zahvaćena aorta i njezini glavni ogranci s odgovarajućom kliničkom slikom (npr. bolnost i neurološki ispadi u području gornjih ili donjih udova i sl.) ili intrakranijski ogranci karotidne arterije (cerebrovaskularna bolest). Prema literaturnim podacima gigantocelularni arteritis može biti uzrokom i do 15 % svih slučajeva vrućice nepoznatoga porijekla kod osoba starijih od 65 godina.
Polimijalgija reumatika je klinički sindrom obilježen bolovima i ukočenošću mišića u području vrata, ramena i zdjeličnoga pojasa, često udružen s vrućicom, slabošću i gubitkom na tjelesnoj težini. Navedene tegobe ograničavaju bolesnike u izvođenju svakodnevnih aktivnosti kao što je oblačenje, češljanje ili sjedanje (ustajanje) sa stolice. Kod manjega broja bolesnika može se zamijetiti i oticanje zglobova, osobito koljena i sternoklavikularnih zglobova.
Dijagnoza se postavlja na temelju anamnestičkih podataka, kliničkoga nalaza, laboratorijskih nalaza, nalaza slikovnih metoda te patohistološkim pregledom uzorka zahvaćene krvne žile.
Laboratorijski nalazi su nespecifični, a obično nalazimo anemiju kronične bolesti (normocitna, normokromna), ubrzanu sedimentaciju eritrocita i povišene markere upale (leukociti, C-reaktivni protein). Česti su i patološki nalazi jetrenih enzima te hipoalbuminemija.
Slikovne metode. Angiografija (CT angiografija, DSA) može prikazati lokalizaciju i opseg stenozirajućih dijelova zahvaćenih arterija, ali ne pokazuje specifična obilježja koja bi se povezivala samo s ovim oblikom vaskulitisa. Danas se sve više primjenjuje i ultrazvučni pregled temporalne arterije koji pokazuje zadebljanje stijenke (tzv. „halo sign“), a ima relativno visoku osjetljivost (75 %) i specifičnost (83 %). PET/CT može biti koristan u otkrivanju vaskulitisa velikih krvnih žila kod bolesnika koji se prezentiraju simptomima polimilagije reumatike (PMR), osobito ako glukokortikoidi nisu doveli do zadovoljavajućega odgovora, kod bolesnika s PMR-om i konstantno povišenim reaktantima upale, te kod bolesnika s PMR-om uz teške konstitucijske simptome.
Patohistološka dijagnostika ključna je za postavljanje konačne dijagnoze. S obzirom na to da je najčešće zahvaćena temporalna arterija, koja je i najdostupnija za biopsiju, najčešće se analiziraju njezini uzorci, a tipični histološki nalazi opisani su gore u tekstu.
Liječenje. Osnovu liječenja čini primjena glukokortikoida (prednizon, metilprednizolon), a u težim oblicima u obzir dolazi i imunomodulatorna terapija.
Glukokortikoidi. Liječenje treba započeti čim prije po postavljanju dijagnoze kako bi se izbjegle komplikacije vezane uz vid, odnosno sljepoća. U tom slučaju liječenje započinje peroralnim predinzonom u dozi od 60 mg/dan. Kod bolesnika koji su razvili sljepoću preporuča se započeti pulsnom kortikosteroidnom terapijom (metilprednizolon u dozi od 1 g/dan kroz tri dana) nakon čega se nastavlja peroralnim prednizonom kako je to opisano. Postotak bolesnika kojima se oporavi vid nakon navedene terapije je nizak. U terapiju se također preporuča uvesti i acetilsalicilna kiselina u nižim dozama (80 - 100 g/dan). Prednizon u dozi od 60 mg/dan treba primjenjivati najmanje mjesec dana prije početka postupnoga snižavanja doze. Duljinu trajanja liječenja i dozu prednizona treba prilagođavati kliničkom stanju i laboratorijskim nalazima (sedimentacija eritrocita, CRP). Postupno smanjivanje doze vrši se za 5 mg svaka dva do tri tjedna dok je bolesnik na dozama većim od 20 mg, a potom za 2,5 mg dok je u dozama između 10 i 20 mg, a potom za 1 mg. Kod većine bolesnika liječenje traje 12 do 18 mjeseci, iako će određeni bolesnici zahtijevati i dugoročnije liječenje. Zbog mogućega razvoja komplikacija dugotrajne terapije kortikosteroidima preporuča se i primjena vitamina D i kalcija te gastroprotekcija.
Imunomodulatorna terapija. Za bolesnike koji ne reagiraju na opisano liječenje u obzir dolazi liječenje metotrekastom i tocilizumabom (inhibitor receptora za interleukin-6).
Prognoza. Uz opisanu terapiju bolesnici načelno imaju dobru prognozu. Ako se razvije sljepoća, ona je obično trajna i tek se kod manjega broja bolesnika ona povlači. Također, kod bolesnika sa zahvaćenom aortom čest je razvoj aneurizmi koje se mogu javiti i desetljećima kasnije (najčešće u prvih 5 do 10 godina).
TAKAYASU ARTERITIS
Definicija i epidemiologija. Takayasu arteritis (bolest bez pulsa, okluzivna trombartropatija) predstavlja granulomatozni vaskulitis nepoznate etiologije koji zahvaća aortu i njezine glavne ogranke, a kod 50 % bolesnika bolest zahvaća i plućnu cirkulaciju. Javlja se obično u mlađoj životnoj dobi (prije četvrtoga desetljeća života) sa značajno većom učestalošću kod žena (odnos 8:1) i to najčešće na području Dalekoga Istoka i Azije.
Etiopatogeneza i patologija. Kao i kod drugih oblika primarnih vaskulitisa, točan uzrok njegovoga nastanka ostaje nepoznat. Kao i kod gigantocelularnoga arteritisa, vjeruje se da patološki proces započinje u adventiciji kao imunološki odgovor na neki nepoznati antigen. Stijenka zahvaćenih arterija prožeta je brojnim limfocitima među kojima dominiraju CD8+ limfociti, dok uloga CD4+ limfocita u ovom obliku vaskulitisa nije istražena i potvrđena. Histološki, ovaj vaskulitis karakterizira razvoj granuloma u ranoj fazi te u kasnijim stadijima postupni razvoj fibroze koja može dovesti do suženja lumena arterija s posljedičnim smanjenim protokom krvi u distalna područja. Najčešća lokalizacija Takayasuovoga arteritisa je luk aorte s njegovim glavnim ograncima (obično potključne arterije). Zahvaćena aorta sklona je kasnijem razvoju aneurizme ili nastanku disekcije.
Klinička slika. Kod većine bolesnika mogu se naći nespecifični, opći simptomi kao što su umor, malaksalost, gubitak na tjelesnoj težini, nejasna febrilna stanja i preznojavanja, a ovisno o zahvaćenim krvnim žilama javljaju se i specifični klinički oblici i manifestacije: koronarne arterije (stenokadija), karotidne arterije (sinkopa, prolazne smetnje vida, retinopatija), potključne arterije (bolovi i neurološki ispadi gornjih udova), renalne arterije (renovaskularna hipertenzija), mezenterične arterije (crijevna ishemija), ilijačne arterije (intermitentna klaudikacija) i sl. Zahvaćanje plućne cirkulacije koje nalazimo kod oko polovine bolesnika rezultira nastankom plućne hipertenzije i opterećenja desnoga srca (cor pulmonale). S obzirom na to da su najčešće zahvaćene potključne arterije i da su posljedično pulsacije na gornjim udovima jedva pipljive, ova bolest se često naziva i bolest bez pulsa (engl. pulseless disease).
Dijagnoza. Postavljanje dijagnoze ovoga vaskulitisa otežano je zbog nedostupnosti uzoraka za patohistološku analizu te široke diferencijalne dijagnoze. Postavljanje dijagnoze temelji se na laboratorijskim i slikovnim radiološkim metodama uz odgovarajuće epidemiološke i kliničke karakteristike (ženski spol, mlađa dob, izraženi opći simptomi). U laboratorijskim nalazima kod većine bolesnika nalazimo anemiju kronične bolesti, ubrzanu sedimentaciju eritrocita i povišene vrijednosti C-reaktivnoga proteina. CT/MR angiografija ili DSA (digitalna suptrakcijska angiografija) mogu ukazivati na stenoze, okluzije ili dilatacije zahvaćenih arterija što podupire dijagnozu ove bolesti.
Liječenje. Temelj liječenja čine glukokortikoidi, a započinje se s peroralnom primjenom prednizona u dozi od 1 mg/kg kroz mjesec dana nakon čega slijedi postupno snižavanje doze kroz nekoliko mjeseci ovisno o kliničkom odgovoru i laboratorijskim nalazima (liječenje nekada može trajati i mjesecima). Korisna je i istodobna primjena acetilsalicilne kiseline. U težim oblicima bolesti ili onima koji ne reagiraju na monoterapiju prednizonom, u terapiju se uključuju i imunoodulatori, kao što su metotreksat, ciklofosfamid, leflunomid, azatioprin ili mofetilmikofenolat. U najtežim oblicima opravdana je i primjena infliksimaba (TNF-α inhibitor) ili tocilizumaba (IL-6 inhibitor). Kirurško liječenje i endovaskularne intervencije mogu biti korisne u rješavanju stenoza ili dilatacija, posebice ako se radi o aorti.
Prognoza. Bolest kod najvećega broja bolesnika pokazuje kroničan i progresivan tijek s čestim relapsima te obično završava nekom od komplikacija (npr. razvoj aneurizme aorte). Razvoj i uključivanje novih lijekova u terapiju smanjuje trend razvoja kroničnih komplikacija.
NODOZNI POLIARTERITIS
Definicija i epidemiologija. Nodozni poliarteritis (PAN) predstavlja nekrotizirajući vaskulitis koji zahvaća srednje velike arterije, najčešće kože, mozga i perifernih živaca (vasa vasorum) te visceralnih organa (koronarne arterije, mezenterijalne arterije, renalne arterije) uz karakterističnu poštedu plućne cirkulacije. Ovaj oblik vaskulitisa češće se javlja kod muškaraca i to obično u srednjoj životnoj dobi.
Etiopatogeneza. PAN predstavlja vaskulitis posredovan imunokompleksima. Nastali imunokompleksi koji se sastoje od antigena i antitijela odlažu se u stijenku arterija te dovode do aktivacije sustava komplementa i posljedičnoga oštećenja. Kod oko 10 % bolesnika pojava PAN-a povezana je s hepatitis B infekcijom, a kod manjega broja oboljelih povezuje se i s infekcijama virusom hepatitisa C te virusom humane imunodeficijencije, a češće se viđa i kod bolesnika s leukemijom vlasastih stanica. Kod ostalih bolesnika predisponirajući antigen ostaje nepoznat. Kod manjega broja bolesnika bolest može biti povezana s mutacijama gena za enzim deaminazu-2, a karakterizira ih rana pojavnost (mlađa životna dob) i pozitivna obiteljska anamneza na PAN.
Patologija. PAN najčešće zahvaća arterije kože, perifernih živaca, mozga i viseceralnih organa. Promjene su segmentalne prirode, najčešće lokalizirane na račvištima. Glavna histološka obilježja uključuju fibrinoidnu nekrozu te pleomorfni stanični infiltrat kojega u najvećem broju čine polimorfonukleari te varijabilan broj limfocita i eozinofila limfocitnu infiltraciju uz odsutnost granuloma. Između zahvaćenih segmenata arterija često nalazimo dilatacijska područja.
Klinička slika PAN obuhvaća opće simptome te simptome vezane uz zahvaćene krvne žile. Opći simptomi uključuju umor, malaksalost, mišićnu slabost, gubitak na tjelesnoj težini te nejasno febrilno stanje. Najčešće specifične manifestacije su kožne, a javljaju se u obliku supkutanih čvorića, ulkusa, gangrene distalnih dijelova prstiju ili kao livedo retikularis. Također, velika većina bolesnika prezentira se i vaskularnom neuropatijom (periferna neuropatija), obično u obliku multiploga mononeuritisa. Ostale manifestacije uključuju mezenterijalnu ishemiju, akutni apendicitis, kolecistitis ili pankreatitis, akutno bubrežno oštećenje (hipertenzija, proteinurija, hematurija), cerebrovaskularni incident, akutni infarkt miokarda i/ili srčano popuštanje.
Dijagnoza se postavlja na osnovi anamnestičkih podataka, kliničkih manifestacija, laboratorijskih nalaza, slikovnih metoda i patohistološke slike zahvaćenih krvnih žila. Laboratorijski nalazi su nespecifični te uključuju anemiju kronične bolesti, leukocitozu, ubrzanu sedimentaciju te povišene vrijednosti C-reaktivnoga proteina. Kod vrlo maloga broja bolesnika ANCA protutijela i/ili reuma faktor mogu biti pozitivni, ali oni nisu specifični za ovaj oblik vaskulitisa. Kod svih bolesnika potrebno je učiniti serološke markere na virusne hepatitise (hepatitis B, hepatitis C). CT ili MR angiografija može ukazivati na segmentalna suženja i poststenotične dilatacije zahvaćenih krvnih žila (CT ili MR angiografija aorte i glavnih ogranaka, cerebralnih arterija i sl.). Patohistološka dijagnostika ključna je za potvrdu dijagnoze (histološki nalaz gore je opisan), a materijal najčešće čini uzorak promjena na koži ili intraoperativno dobiven uzorak.
Liječenje PAN kod većine bolesnika započinje peroralnom primjenom prednizolona u dozi od 60 mg/dan uz kasnije postupno snižavanje ovisno o kliničkom odgovoru i laboratorijim nalazima. Kod bolesnika koji imaju znakove životne ugroženosti (istovremena zahvaćenost više krvnih žila) u liječenju se primjenjuje i pulsna kortikosteroidna terapija (1 g metilprednizolona kroz tri dana). Kao druga linija liječenja kod bolesnika koji ne reagiraju na glukokortikoide, i/ili imaju teža klinička očitovanja, indicirana je primjena ciklofosfamida (2 mg/kg/dan pero os ili 15 mg/kg/mjesec intravenski) koji se primjenjuje kroz 6 do 12 mjeseci, a potom se prelazi na terapiju održavanja uz azatioprin, metotreksat li leflunomid. Za bolesnike koji imaju PAN povezan za hepatitis B infekcijom preporučeno je liječenje plazmaferezom. U liječenju PAN bolesnika s mutacijom gena za deaminazu 2 koriste se TNF-α inhibitori.
Prognoza neliječenih bolesnika je loša i petogodišnje preživljenje se kreće oko svega 10 %. Uz zadovoljavajuće liječenje i redovne kontrole taj se postotak povećava na 60 do 90 %. Za razliku od ostalih oblika vaskulitisa nakon jednom izazvane remisije, relapsi bolesti su rijetki i mogu se očekivati kod oko 20 % oboljelih.
OSTALI VASKULITISI SREDNJE VELIKIH KRVNIH ŽILA
Kawasakijeva bolest označava arteritis malih i srednje velikih krvnih žila udružen s promjenama na sluznici i s povećanim limfnim čvorovima. Javlja se kod djece pa se opisuje u sklopu pedijatrije. Uzrok je nepoznat, vjerojatno se radi o imunološkom mehanizmu. Može zahvatiti i koronarne krne žile i uzrokovati miokardijalnu ishemiju. Burgerova bolest opisana je u sklopu Kardiologije (Ostale bolesti perifernih arterija).
GRANULOMATOZA S POLIANGIITISOM (WEGENEROVA GRANULOMATOZA)
Granulomatoza s poliangiitisom (GPA) ili Wegenerova granulomatoza predstavlja kroničnu autoimunu bolest nepoznate etiologije koju karakterizira granulomatozna upala gornjih dišnih puteva i pluća, nekrotizirajući vaskulitis malih i srednje velikih krvnih žila te fokalni glomerulonefritis. Detaljnije o etiopatogenezi, kliničkoj slici, dijagnostici i liječenju pisano je u poglavlju Granulomatozne intersticijske bolesti pluća (Pulmologija).
EOZINOFILNA GRANULOMATOZA S POLIANGIITISOM (MORBUS CHURG-STRAUSS)
Eozinofilna granulomatoza s poliangiitisom ili Churg-Straussov sindrom je rijetka imunološki posredovana sistemska bolest koju karakterizira nekrotizirajući ANCA-pozitivni vaskulitis te stvaranje granuloma i to najčešće u plućnom intersticiju, iako bolest može zahvatiti i druge organe i tkiva. Detaljnije o etiopatogenezi, kliničkoj slici, dijagnostici i liječenju opisano je u poglavlju Granulomatozne intersticijske bolesti pluća (Pulmologija).
MIKROSKOPSKI POLIANGIITIS
Definicija. Mikroskopski poliangiitis (MPA) je pauci-imuni negranulomatozni nekrotizirajući vaskulitis kojeg karakterizira: 1) zahvaćanje arterija, vena i kapilara; 2) sklonost plućnoj i bubrežnoj vaskulaturi te 3) prisutnost ANCA protutijela. Bolest se obično javlja između trećega i petoga desetljeća života, podjednako u oba spola.
Etiopatogeneza MPA-e nije poznata, ali pokazuje određenu sličnost prethodno opisanome nodoznom poliarteritisu (PAN). Glavne razlike su u tome što su kod bolesnika s MPA-om obično zahvaćena pluća u odnosu na one s PAN-om, a MPA obično nije povezan s prethodnom hepatitis B ili C infekcijom kao što je to slučaj kod PAN-a. U rijetkim slučajevima primjena nekih lijekova kao što su alopurinol, hidralazin, propiltiouracil, penicilamin te sulfasalazin može biti povezana s razvojem MPA-e. MPA najčešće pogađa bubrege i pluća pa gotovo kod svih bolesnika nalazimo brzoprogresivni glomerulonefritis (90 - 100 %), dok difuznu alveolarnu hemoragiju nalazimo kod 30 %, a pulmonalne infiltrate do 50 % bolesnika.
Klinička slika. MPA uključuje široki spektar kliničkih manifestacija s obzirom na to da može zahvatiti brojne i različite krvne žile, a u svom kliničkom tijeku nalikuje na PAN. Gotovo kod svih bolesnika s MPA-om nalazimo znakove oštećene funkcije bubrega (brzoprogresivni glomerulonefritis) s ili bez simptoma i znakova zahvaćenosti pluća (difuzna alevolarna hemoragija) te sam MPA uz Goodpastureov sindrom danas predstavlja jedan od najčešćih uzroka pulmorenalnoga sidnroma. Pored toga se mogu javiti i opći simptomi te simptomi od strane zahvaćenosti drugih organa i tkiva (kožne lezije, periferna neuropatija, gastrointestinalna krvarenja, artralgije i dr.).
Dijagnoza se postavlja na temelju anamnestičkih podataka i laboratorijskih nalaza, a potvrđuje se patohistološkom analizom zahvaćenoga tkiva. U laboratoriju nalazimo anemiju kronične bolesti, povišene upalne parametre, patološki nalaz urina (proteinurija, eritrociturija, eritrocitni cilindri) te pozitivna ANCA antitijela (kod 50 do 60 % bolesnika radi se o p-ANCA, dok kod 15 do 30 % bolesnika nalazimo c-ANCA antitijela). Histološki nalaz uključuje fokalni, segmentirajući nekrotizirajući vaskulitis s upalnim mješovitim infiltratom, bez formiranja granuloma na svjetlosnoj mikroskopiji, dok pregledom elektronskim mikroskopom ne nalazimo imunodepozite. Najčešći bioptički uzorak je bioptat kožne promjene ili bubrega.
Liječenje i prognoza. Liječenje MPA-e isto je kao i kod bolesnika s granulomatozom i poliangiitisom (gore opisano), najčešće kombinacijom glukokortikoida, ciklofosfamida ili rituksimaba. Plazmafereza i IVIG korišteni su kod nekoliko pacijenata s progresivnim bubrežnim zatajenjem i plućnom hemoragijom. Prognoza bolesnika ponajprije ovisi o vremenu postavljanja dijagnoze i početku liječenja s obzirom na brzi razvoj fibroze i kronične bubrežne bolesti.
BOLEST GLOMERULARNE BAZALNE MEMBRANE (GOODPASTUREOV SINDROM)
Definicija i epidemiologija. Bolest glomerularne bazalne membrane ili Goodpastureov sindrom predstavlja autoimunu bolest pri kojoj nastaju autoantitijela na kolagen tipa IV (α3-domena) kojega nalazimo u sastavu glomerularne bazalne membrane te u području alvelokapilarne stijenke zbog čega dolazi do razvoja renopulmonalnoga sindroma kojega čine brzoprogresivni glomerulonefritis i difuzna alveolarna hemoragija. Pojavnost ove bolesti pokazuje karakterističnu dobnu bimodalnu pojavnost pri čemu se ona najčešće susreće u dobi između 20. i 30. godine života te između 60. i 70. godine života. U mlađoj životnoj dobi bolest se češće javlja u muškaraca, dok je u starijoj dobi češća kod žena.
Etiopatogeneza. Bolest nastaje kao posljedica stvaranja autoantitijela na α3-domenu kolagena tipa IV (Goodpastureov antigen) koji se u najvećoj koncentraciji nalazi u glomerularnoj bazalnoj membrani te alvelokapilarnoj stijenci. Točan mehanizam nastanka ovih autoantitijela nije razjašnjen, ali se zna da ulogu ima genetska predispozicija (HLA-DR15 i HLA-DR4 haplotipovi) te izloženost organskim otapalima (ugljikovodici), metalnoj prašini, nikotinu (pušenje), infekcijama (posebno influenza) te nekim lijekovima (alemtuzumab). Navedeni rizični čimbenici dovode do „otkrivanja“ antigena što omogućuje stvaranje i interakciju s autoantitijelima pri čemu se formiraju imunokompleksi i aktivira sustav komplementa koji dovodi do oštećenja tkiva (reakcija preosjetljivosti tipa II). Stvorena autoantitijela su pretežno IgG klase (IgG1 i IgG3 subklasa), rjeđe IgA i IgM.
Klinička slika. Najveći broj bolesnika prezentira se kao renopulmonalni sindrom (60 - 80 %), dok se manji dio prezentira samo kao izolirana bubrežna bolest (20 - 40 %) ili plućna bolest (5 - 10 %). Kliničku sliku čine opći, plućni i bubrežni simptomi. Opći simptomi uključuju slabost, malaksalost, vrućicu, algične sindrome te anoreksiju. Oni se mogu javiti prije ili usporedo s plućnim i bubrežnim manifestacijama. Plućnu simptomatologiju označavaju kašalj, dispneja i hemoptize (masivna plućna hemoragija je rijetka, ali po život opasna manifestacija). Bubrežne manifestacije obilježavaju oligurija, hematurija, arterijska hipertenzija, edemi te razvoj uremijskoga sindroma. Bubrežna simptomatologija i propadanje bubrežne funkcije odvija se kroz nekoliko tjedana do nekoliko mjeseci (najčešće u roku od jednoga do dva tjedna).
Dijagnoza se postavlja na temelju kliničke slike (akutno bubrežno oštećenje, hemoptize, dispneja), laboratorijskih nalaza, radiološkoga nalaza pluća te nalaza biopsije bubrega. U laboratorijskim nalazima obično nalazimo anemiju, leukocitozu, ubrzanu sedimentaciju eritrocita, povišene dušične metabolite te patološki nalaz urina (proteinurija nefrotskoga ranga, eritrociturija, eritrocitni cilindri). Glavni serološki biljeg kod ovih bolesnika su pozitivna anti-GBM protutijela koja imaju visoku osjetljivost (> 95 %) i specifičnost (> 97 %), a kod oko jedne trećine bolesnika mogu se naći i pozitivna ANCA autoantitijela. Radiološka dijagnostika pluća (klasični rendgenogram, HRCT) pokazuje obostrane mekotkivne infiltrate koji s napredovanjem bolesti poprimaju intersticijski uzorak. Biopsija bubrega i patohistološka analiza uzorka daju nam konačnu dijagnozu koja se temelji na nalazu svjetlosne mikroskopije (polumjeseci) i imunofluorescencije (linearni IgG depoziti).
Liječenje. Početno liječenje čini imunosupresivno liječenje temeljeno na glukokortikoidima s ili bez dodatka ciklofosfamida te primjenom postupka plazmafereze. Obično se počinje s pulsnom primjenom metilprednizolona (500 - 1000 mg intravenski kroz tri dana) nakon čega se nastavlja peroralnim prednizolonom u dozi od 1 mg/kg (maksimalno 60 - 80 mg/dan) do postizanja remisije kada se postupno snižava doza. Ciklofosfaid primjenjuje se u dozi od 1 do 2 mg/kg/dan. Liječenje obično traje 3 do 6 mjeseci, a nekada i dulje.
Prognoza ponajprije ovisi o vremenu početka liječenja. Bolest karakterizira brzo propadanje bubrežne funkcije te, ako se bolesnici počnu specifično liječiti u trenu kada već zahtijevaju hemodijalizu, rijetko je zabilježen povrat bubrežne funkcije. Relapsi bolesti su rijetki. Dominantna ugroženost bolesnika proizlazi iz difuzne alveolarne hemoragije i nastanka akutnoga respiratornog distres sindroma. Ukupno petogodišnje preživljenje kreće se oko 80 %.
IgA VASKULITIS (HENOCH-SCHONLEINOVA PURPURA)
Definicija. IgA vaskulitis ili Henoch-Schonlein purpura (HSP) predstavlja vaskulitis malih krvnih žila koji nastaje uslijed odlaganja imunokompleksa koji sadrže imunoglobulin A u stijenku krvne žile, a dominantno se javlja kod djece i mlađih odraslih osoba, iako se može javiti u svakoj životnoj dobi (odrasli imaju teži oblik bolesti s većom učestalosti zahvaćanja bubrega). Osnovna obilježja HSP-a su palpabilna purpura, artalgije, probavne smetnje (akutni abdomen) te glomerulonefritis.
Etiopatogeneza. Točan uzrok nastanka i mehanizam bolesti nije poznat. Glavni i ključni događaj u patogenetskom smislu je odlaganje imunokompleksa u područje stijenke krvne žile. Oni se sastoje od molekula IgA i protutijela usmjerenih na njih (IgA1-IgG imunokompeksi). Vjeruje se da ti imunokompleksi nastaju kod osoba koji imaju autosomno-dominantni nasljedni poremećaj glikozilacije IgA pri čemu nastaju uvjetno patogene molekule IgA na koje mogu križno reagirati protutijela nastala tijekom infekcija ili nakon primjene cjepiva, lijekova ili nekih drugih antigena (toksini insekata i sl.). Istaloženi imunokompleksi u stijenkama krvnih žila imaju sposobnost aktivacije sustava komplementa alternativnim putem što dovodi do nastanka upale i oštećenja. Osim u krvne žile, nastali imunokompeksi talože se i u mezangij glomerula što uvjetuje nastanak IgA nefropatije.
Klinička slika. Uz opće simptome koji uključuju umor, febrilno stanje, gubitak teka i artralgije, javljaju se i specifična klinička očitovanja vezana uz zahvaćene krvne žile, a najčešće su zahvaćeni koža, probavni sustav, bubrezi i sinovija zbog čega se kod više od 80 % bolesnika javlja karakteristični trijas: palpabilna purpura, bol u trbuhu i artritis. Kod odraslih je bolesnika pojava bolova u trbuhu rjeđa pojava, ali se zato češće susreće IgA nefropatija. Osim palpabilne purpure, kožne promjene mogu uključivati i petehije, ekhimoze te makulo-papulozni osip, a kožne promjene najizraženije su u području donjih udova. Artritis dominantno zahvaća koljena i gleženjeve pri čemu su oni bolni, otečeni i crveni. Kod trećine bolesnika razvijaju se difuzni, grčeviti bolovi u trbuhu praćeni mučninom i povraćanjem što nekada može nalikovati i na akutno kirurško zbivanje u trbuhu. Probavne manifestacije posljedica su edema i submukoznoga krvarenja unutar probavne cijevi (najčešće jejunuma). Simptomi IgA nefropatije opisani su u poglavlju Glomerularne bolesti bubrega. Rjeđa klinička očitovanja mogu uključivati i središnji živčani sustav (glavobolja, konfuzija, konvulzije) s mogućim razvojem posteriorne reverzibilne encefalopatije (PRES), intrakranijskoga krvarenja ili ishemijskoga moždanog udara (ovo se opisuje kod manje od 7 % bolesnika). Zahvaćenost pluća opisana je samo u rijetkim, pojedinačnim slučajevima.
Dijagnoza se postavlja na temelju kliničke slike, laboratorijskih nalaza, specifičnih slikovnih metoda (ultrazvuka abdomena, obojenog doplera, angiografije), a potvrđuje se patohistološki. Nespecifični laboratorijski nalazi ukazuju na povišene vrijednosti upalnih parametara i ubrzanu sedimentaciju ertirocita, a specifično nalazimo povišenu razinu IgA1 te povišeni titar IgA1-IgG (ili rjeđe IgA1-IgA) imunokompleksa uz većinom uredne vrijednosti komponenata komplemnta (C3 i C4). Kod IgA nefropatije nalazimo i povišene vrijednosti metabolita dušika te patološki nalaz urina. Konačna dijagnoza se postavlja patohistološkim pregledom bioptata kožnih promjena ili bubrega (ako nalazimo znakove bubrežnoga oštećenja). Histološki nalaz kožnih promjena uključuje leukocitoklastični vaskulitis s depozitima IgA i komponente C3 u stijenci krvnih žila. Pacijenti za koje se sumnja da imaju IgAV i čija je biopsija negativna za IgA trebaju se dodatno obraditi zbog diferencijalne dijagnoze na ANCA vaskulitis, antiC1q bolest i IgA paraproteinemiju.
Liječenje. Osnovu liječenja čine glukokortikoidi koji često ne pokazuju zadovoljavajući učinak kod ovih bolesnika. U blažim oblicima primjenjuje se peroralno perdnizon u dozi do 1 mg/kg (maksimalno 60 - 80 mg/dan) dok u težim oblicima u obzir dolazi i primjena pulsne glukokortikoidne terapije (metilprednizolon 500 - 1000 mg/dan kroz tri dana). Kod bolesnika koji ne reagiraju u obzir dolazi primjena i drugih imunosupresiva (azatioprin, ciklofosfamid, mikofenolat). Kod djece je bolest obično samoograničavajuća i spontano prolazi kroz nekoliko tjedana, dok kod odraslih, kod kojih se češće javlja zahvaćenost bubrega (IgA nefropatija), zaostaju i trajne posljedice u obliku kronične bubrežne bolesti.
KRIOGLOBULINEMIJA I KRIOGLOBULINEMIČNI VASKULITIS
Krioglobulinemija je naziv za prisutnost cirkulirajućih krioglobulina u serumu bolesnika, a može biti povezana s razvojem hiperviskoznoga sindroma i vaskulitisa. Krioglobulini su imunoglobulini koji imaju sposobnost precipitacije i taloženja na temperaturama ispod 37℃ i ponovnoga otapanja kada se izlože višim temperaturama. U načelu razlikujemo tri tipa krioglobulina s obzirom na njihov sastav. Tip I krioglobulina čine monoklonalni imunoglobulini (najčešće IgM) koji se mogu vidjeti u sklopu hematoloških malignih oboljenja i koji su povezani s nastankom hiperviskoznoga sindroma. Tip II krioglobulina sastoji se od monoklonalnoga IgM i poliklonalnoga IgG imunoglobulina, dok se tip III sastoji od poliklonalnoga IgM i poliklonalnoga IgG imunoglobulina, a njihova se pojavnost viđa pri različitim sistemskim bolestima vezivnoga tkiva i nekih infekcija (hepatitis C), a povezani su s nastankom vaskulitisa. U više od 90 % slučajeva krioglobulinemije uzrok je poznat (limfoproliferativno oboljenje, autoimuna bolest, hepatitis C, infektivni endokarditis), dok se u ostalih 10 % slučajeva ne zna točan uzrok nastanka krioglobulina (idiopatski oblik).
Krioglobulinemični vaskulitis je rijedak oblik vaskulitisa koji zahvaća male i srednje velike krvne žile, a povezan je s odlaganjem imunokompleksa koji sadrže krioglobuline u stijenku krvnih žila gdje potom aktiviraju sustav komplementa koji dovodi do oštećenja zahvaćenih tkiva i organa. Najčešće kliničke manifestacije ovoga oblika vaskulitisa uključuju recidivirajuću palpabilnu purpuru i perifernu neuropatiju, a kod nekih bolesnika moguć je i razvoj brzoprogresivnoga glomerulonefritisa (osobito kod bolesnika s krioglobulinemijom tip II). Ostale kliničke manifestacije uključuju gangrenu okrajina (prsti šaka i stopala), mezenterijalnu ishemiju i sl. Dijagnoza se postavlja na temelju kliničke slike, anamnestičkih podataka i pozitivnoga serumskog testu na krioglobuline. Obično nalazimo i sniženu vrijednost C4 komponente komplementa. Liječenje se temelji na liječenju samoga uzroka krioglobulinemije, a kod pojave vaskulitisa primjenjuje se imunosupresivno liječenje (glukokortikoidi, ciklofosfamid, azatioprin, metotreksat, ciklosporin, kolhicin, rituksimab), a dodatnu korist može pružiti i terapijska plazmafereza te intravenski humani imunoglobulini.
HIPOKOMPLEMENTARNI URTIKARIJSKI VASKULITIS
Hipokomplemetarni urtikarijski vaskulitis rijedak je oblik vaskulitisa koji se obično javlja kod bolesnika koji već od ranije imaju sistemsku autoimunu bolest (sistemski eritematozni lupus, Sjögrenov sindrom), a obilježen je stvaranjem autoantitijela na kolagenu sličnu regiju komponentne komplementa C1q. Tako nastaju tzv. C1q precipitati koji se sastoje od autoantitijela klase IgG na navedeni antigen koji se odlažu u stijenku kožnih krvnih žila te dovode do aktivacije sustava komplementa. Iako su kožne lezije koje nastaju pri ovom obliku vaskulitisa (a koje predstavljaju i temeljnu kliničku manifestaciju) nalik onima kod urtikarije, nekoliko je bitnih razlika: promjene uvjetovane vaskulitisom traju dulje (> 48 sati), obično imaju purpurnu komponentnu i nakon njihova povlačenja zaostaju postupalne hiperpigmentacije. Osim toga, kožne lezije su bolne (za razliku od prave urtikarije gdje svrbe), veličine su od 0.5 to 5 cm u promjeru (dok kod prave urtikarije mogu biti veće od 10 cm) te obično zahvaćaju trup i proksimalne dijelove ekstremiteta. Kod ovih bolesnika mogu se naći i artritis, glomerulefritis te znakovi zahvaćanja pluća i očiju. Dijagnoza se postavlja na temelju specifičnih laboratorijskih nalaza (snižene vrijednosti C1q te snižene ili uredne vrijednosti C3 i C4 komponente, povišeni titar anti-C1q te kod 60 % bolesnika pozitivna antinuklearna antitijela i kod 10 % bolesnika reumatoidni faktor) te patohistološke analize kožnih lezija (leukocitoklastični vaskulitis koji zahvaća kapilare i postkapilarne venule s depozitima imunoglobulina i komponentni komplementa u i oko stijenke krvnih žila). Specifični oblici liječenja ne postoje, a od koristi mogu biti kortikosteroidi i imunosupresivi, a što ponajprije ovisi o težini kliničke slike (prednizon, hidroksiklorokin, dapson). Uspješnost terapije odražava pad vrijednosti titra anti-C1q uz normalizaciju C3 i C4, dok vrijednost C1q kod većine bolesnika ostaje trajno snižena. Prognoza ovih bolesnika u prvom redu ovisi o težini kliničke slike i zahvaćenosti krvnih žila.
BEHCETOV SINDROM
Definicija i epidemiologija. Behcetova bolest predstavlja rijedak oblik vaskulitisa koji je karakteriziran rekurentnim aftoznim stomatitisom, uveitisom, genitalnim ulceracijama i kožnim lezijama. Pojavnost bolesti je različita u raznim geografskim područjima tako da se široko kreće, od 1:10 000 u Japanu do 1:500 000 u Europi i Sjevernoj Americi. Spolna pojavnost bolesti vezana je uz geografska područja tako da se bolest u zemljama Srednjeg istoka (geografsko područje Puta svile) češće javlja kod muškaraca (odnos muškarac:žena je 3-5:1), dok se u Europi i Americi češće javlja kod žena (odnos žena:muškarac je 5:1). Muškarci obično imaju teže oblike bolesti od žena. Bolest se javlja pretežno u mlađoj životnoj dobi.
Etiopatogeneza. Iako etiologija i patogeneza Behcetove bolesti nije razjašnjena, danas se smatra je ona povezana s genetskom predispozicijom te poremećenim imunološkim odgovorom na zasada nepoznati antigen (okolišni čimbenik). O Behcetovoj bolesti najčešće se govori kao o vaskulitisu autoimune etiologije, iako određene karakteristike ne govore u prilog klasičnoj autoimunoj bolesti: češća je pojavnost kod muškaraca nego kod žena, nije povezana i ne dolazi s drugim autoimunim bolestima te nije povezana s povećanom učestalosti HLA-DR3 i HLA-DR4 antigena kao što je to slučaj u drugim autoimunim bolestima.
Genetika. Iako je Behcetova bolest sporadična bolest, zabilježeno je i obiteljsko pojavljivanje što zajedno s geografskom i rasnom rasprostranjenosti ukazuje na genetsku predispoziciju pojavnosti bolesti. Nositelji HLA-B51 i HLA-B5 antigena imaju povećan rizik od pojavnosti Behcetove bolesti od onih koji ne posjeduju navedene antigene, ososbito u endemskom području. S obzirom na to da je obiteljsko pojavljivanje Behcetove bolesti rijetko, njezina patogeneza ne može se objasniti samo genetskim čimbenicima, pa se i pretpostavlja da u pojavljivanju bolesti veliki dio imaju i vanjski, okolišni čimbenici.
Okolišni čimbenici. Danas se najviše govori o infektivnim uzročnicima kao najuvjerljivijim vanjskim čimbenicima koji mogu biti povezani s nastankom Behcetove bolesti. Kao najizgledniji, navodi se herpes simpleks virus tip I (HSV1) čiji je genom identificiran u perifernoj krvi, limfocitima i monocitima većine bolesnika s Behcetovom bolesti. Drugi infektivni agensi koje se spominju kao mogući kandidati su virusi hepatitisa B i C, parvovirus B19, streptokoki, mikobakterije, Borellia burgdorferi, Helicobacter pylori te Saccharomyces cerevisiae.
Imunopatofiziologija. Iako točan slijed događaja koji dovodi do oštećenja stanica i tkiva u Behcetovoj bolesti nije poznat, predloženi mehanizam stavlja u prvi plan hipersenzibilizaciju T-limfocita za koje se smatra da su pokretači cijeloga mehanizma. Aktivacija T-limfocita nastaje interakcijom s AP-stanicama (predočuju antigen) putem TCR receptora (engl. T-cell antigen receptor), iako još nije jasno koji su antigeni odgovorni za njihovu aktivaciju. Sezibilizirani T-limfociti aktiviraju monocite interakcijom molekula CD40-CD150, a aktivirani monociti, kao i same T-stanice luče brojne citokine kao što su TNF-α, IFN-γ, IL-1, IL-8 IL 12 te IL-17 koji vode prema Th1 obrascu imunološkoga odgovora. Svi navedeni citokini djeluju na aktivaciju makrofaga, neutrofilnu kemotaksiju i fagocitozu koji konačno dovode do oštećenja ciljnih stanica i tkiva. Hiperfunkcija neutrofila (formiranje neutrofilne ekstracelularne mreže, engl. neutrophil extra-cellular traps, odnosno tzv. „NEToza“) uključuje pojačanu kemotaksiju, produkciju slobodnih kisikovih radikala, fagocitozu te tkivnu infilitraciju i ozljedu tkiva.
Patologija. Glavna histopatološka promjena koja se nalazi u podlozi kliničkih manifestacija Behcetove bolesti je vaskulitis koji dominantno zahvaća vene, ali može zahvatiti i arterije. Kožne lezije karakterizira neutrofilna vaskularna reakcija ili potpuno razvijen leukocitoklastični vaskulitis. Mikroskopski, kapilare i venule dermisa pokazuju neutrofilne infiltrate, ekstravazaciju eritrocita s ili bez fibrinoidne nekroze.
Klinička slika. Behcetova bolest je multisistemska bolest koja zahvaća različita tkiva i organe kao što koža, sluznice, oči, zglobovi te male krvne žile različitih organa, najčešće probavnoga, mokraćnoga i središnjega živčanog sustava. Kao i kod svih drugih sistemskih bolesti, kod bolesnika nalazimo i opće simptome kao što su opća slabost i malaksalost, brzo umaranje, nejasna febrilna stanja te gubitak teka i posljedični gubitak na tjelesnoj težini. Najčešće specifične manifestacije ove bolesti uključuju oralne, genitalne, očne te kožne promjene.
Kod više od 95 % oboljelih nalazimo oralne promjene u obliku aftoznih ili herpetiforminih ulceracija koje mogu biti lokalizirane bilo gdje u usnoj šupljini, a mogu se javljati pojedinačno ili u nakupinama i imati različit izražaj (manji ili veći ulkusi). Česti recidivi i povoljan učinak pušenja i cigaretnoga dima na ove promjene govore u prilog Behcetove bolesti. Slične promjene se kod 60 do 80 % bolesnika mogu naći i u genitalnoj regiji (skrotum i prepucij u muškaraca te labije i vagina kod žena). Genitalne ulceracije su bolnije od oralnih te cijele uz stvaranje ožiljaka. Kožne promjene također se mogu vidjeti kod velikoga broja bolesnika, a uključuju papulopustulozne ili akneiformne promjene koje se mogu javiti na bilo kojemu dijelu tijela. Kod ovih bolesnika karakteristična je i nespecifična dermalna upalna reakcija na ogrebotinu ili intradermalno injiciranje fiziološke otopine (patergijski test) koja se očituje pojavom eritematozne papule ili pustule do 48 sati od traume. Očne promjene obično nastupaju dvije do tri godine od pojave same bolesti, a mogu se naći kod 40 do 70 % oboljelih. Prednji uveitis je najčešća manifestacija, dok se stražnji uveitis i hipopion (koji se inače smatra karakterističnim znakom Behcetove bolesti) znatno rjeđe susreće, ali ima nepovoljniji klinički tijek. Rijetko se mogu javiti i optički neuritis, okluzija retinalne arterije, ablacija retine te u konačnici gubitak vida.
Ostale, rjeđe kliničke manifestacije Behcetove bolesti uključuju artritis i artralgije (neerozivni, nedeformirajući oligoartritis koji zahvaća koljena, gležnjeve ili ručne zglobove i prolazne je naravi); vaskularni poremećaji (duboka venska tromboza, tromboza portalne i hepatalne vene, troboza venskih sinusa tvrde moždane ovojnice, formiranje arterijskih aneurizmi); gastrointestinalni poremećaji (anoreksija, povraćanje, dispepsija, dijareja, abdominalne kolike, krvarenje); bubrežni i mokraćni poremećaji (nepsecifični nefritis, IgA nefropatija, neurogeni mokraćni mjehur) te poremećaji središnjega živčanoga sustava (glavobolja, hemipareza, hemiplegija, promjene ponašanja i sl.).
Dijagnoza Behcetove bolesti temelji se na sintezi kliničkih manifestacija, laboratorijskih nalaza kao i slikovnih i histopatoloških nalaza. Laboratorijski nalazi su nespecifični te se mogu naći anemija kronične bolesti, ubrzana sedimentacija eritrocita, leukocita te povišeni C-reaktivni protein. Serumski imunoglobulini mogu, ali ne moraju biti povišeni, dok su autoantitijela kao što su reumatoidni faktor, antinuklearna antitijela, antineutrofilna citoplazmatska antitijela negativna. Slikovne dijagnostičke metode koristimo ovisno o indikaciji i kliničkoj prezentaciji bolesti (radiografsko snimanje lokomotornoga sustava, CT ili MR angiografija i sl.). Temelj dijagnoze čini biopsija patološkoga žarišta, odnosno lezije (najčešće kože, ali može i unutarnjih organa kao što su bubrezi ili organi probavne cijevi). Glavni patohistološki nalaz uključuje vaskulitis s dominantnom limfo-monocitnom infiltracijom te žarišnom nekrozom okoline krvnih žila. U samoj dijagnostici Behcetove bolesti koriste se ISG dijagnostički kriteriji koji su niže opisani.
ISG dijagnostički kriteriji. Inicijalni ili uključni kriterij za postavljanje ove dijagnoze je pojava minimalno tri epizode oralnih aftoznih ili herpetiformnih ulceracija u zadnjih 12 mjeseci (potvrđeno od strane liječnika ili prema navodu bolesnika), a da bi se dijagnoza i potvrdila, potrebno je pronaći još najmanje dva od sljedećih kriterija: 1) rekurentni, bolni genitalni ulkusi koji cijele uz ožiljke; 2) oftalmičke manifestacije, uključujući prednji ili stražni uveitis, hipopio ili retinalni vaskulitis; 3) kožne lezije koje mogu biti nalik na nodozni eritem ili se radi o papulo-pustuloznim promjenama te 4) pozitivan patergijski kožni test: formiranje sterilne eritematozne papule veličine 2 mm ili više u dijametru, koja se pojavljuje 48 sati nakon zagrebavanja kože sterilnom iglom. Osim navedenih kriterija, u obzir treba uzeti i sljedeće karakteristike: višeorganska zahvaćenost, pojavnost bolesti u mlađoj životnoj dobi (najčešće između 25. i 35. godine života) te tijek bolesti obilježen egzacerbacijama i relapsima.
Liječenje. Kod blagih i umjerenih oblika bolesti koji se prvenstveno prezentiraju promjenama na koži i sluznicama, najčešće se koriste niske doze glukortikoida, iako se mogu koristiti i drugi lijekovi, kao što su kolhicin, dapson, azatioprin te apremilast. Kod bolesnika s težim oblicima bolesti u liječenju se prvenstveno primjenjuju visoke doze glukokortikoida (prednizon 1 mg/kg/dan), a u nekim slučajevima i drugi imunosupresivni i imunomodulatorni lijekovi kao što su azatioprin, ciklosporin i metotreksat. U najtežim oblicima bolesti, a osobito kod zahvaćenosti oka i središnjega živčanog sustava koji ne reagiraju na navedene metode liječenja indicira se primjena ciklofosfamida, TNF-α inhibitora (kao što su infliksimab ili adalimumab) te tocilizumaba.
KOŽNI LEUKOCITOKLASTIČNI VASKULITIS
Kožni leukocitoklastični vaskulitis ili kožni leukocitoklastični angiitis je oblik vaskulitisa uzrokovan odlaganjem imunokompleksa u stijenke krvnih žila kože koji nastaju u tijeku infekcija ili reakcija preosjetljivosti na lijekove. Kod većine bolesnika izostaju sistemski simptomi i znaci, iako se kod nekih mogu naći znaci sinovitisa ili vrućica. Kožne promjene slične su purpuri, distribuirane po koži donjih ekstremiteta i gluteusima, a mogu biti asimptomatske ili praćene osjećajem pečenja i svrbeža. Promjene se javljaju 10 do 14 dana od izlaganja lijeka ili početka infekcije. Dijagnoza se postavlja na osnovi kliničkih karakteristika i nalaza biopsije kože (neutrofilna infiltracija u području stijenke krvnih žila i oko njih, fibrinoidna nekroza, disrupcija stineke krvnih žila i ekstravazacija eritrocita). Liječenje se temelji na etiologiji (liječenje infekcije, izostanak lijeka), a kod blažih oblika mogu biti dovoljni antihistaminici (blokatori histaminskih H1 receptora) s ili bez prednizona u malim dozama. U težim oblicima primjenjuje se kolhicin, hidroksiklorokin i dapson, a visoke doze glukokortikoida rezervirane su za bolesnike s najtežim oblicima bolesti (ulceracije, gangrena).
PRIMARNI VASKULITISI SREDIŠNJEGA ŽIVČANOG SUSTAVA
Uvod. Primarni angiitis središnjega živčanog sustava (PACNS) naziv je za vaskulitis koji isključivo pogađa male i srednje velike krvne žile mozga i kralježične moždine, a klinički se preklapa s reverzibilnim cerebralnim vazokostrikcijskim sindromom (RCVS) i često se zamijeni za njega. Etiopatogeneza u oba slučaja je nepoznata i klinički fokus usmjeren je na kliničku sliku, dijagnostiku i liječenje.
Primarni angiitis središnjega živčanog sustava (PACNS) klinički se prezentira širokim dijapazonom neuroloških manifestacija koje mogu uključivati različiti stupanj glavobolje, kvalitativne i kvantitativne poremećaje stanja svijesti, žarišne neurološke ispade, konvulzivne atake uz odsutne opće (sistemske) simptome koje nalazimo u drugim oblicima vaskulitisa. Klinička slika ovih bolesnika postupno se razvija i progredira, za razliku od RCVS-a pri kojemu je početak bolesti nagliji i dramatičniji. Također, laboratorijski nalazi u ovih bolesnika uglavnom su uredni. U uzorku cerebrospinalne tekućine dobivene lumbalnom punkcijom obično se nađe limfocitna pleocitoza i povišena razina proteina. Nalaz MR mozga i MR angiografije krvnih žila mozga može biti uredan, ali obično MR mozga ukazuje na zone ishemije i hemoragizacije u područjima zahvaćenih krvnih žila, a MR angiografija najčešće pokazuje tzv. nalaz krunice u kojemu zahvaćena krvna žila pokazuje segmentalna područja suženja i mjesta dilatacije između njih. Ipak, ovaj nalaz nije patognomoničan za PACNS jer se može naći i kod bolesnika s RCVS-om pa se i dijagnoza često temelji na isključenju ostalih potencijalnih uzroka (infekcije, neoplazme, metabolički poremećaji) te histološkom nalazu bioptata moždanoga tkiva. Liječenje PACNS-a temelji se na primjeni prednizona i ciklofosfamida kroz 6 do 12 mjeseci, a odgovor i prognoza povezani su s veličinom arterija koje su zahvaćene (zahvaćenost malih, kortikalnih i leptomeningealnih arterija povezani su s boljim terapijskim odgovorom i prognozom).
Reverzibilni cerebralni vazokonstrikcijski sindrom (RCVS) znatno se češće susreće nego PACNS, i to s drastično većom učestalošću kod ženskoga spola (80 % bolesnika su žene). Pri kliničkom razlikovanju RCVS-a od PACNS-a najbitniju značajku ima početak bolesti koje je kod RCVS-a nagliji i izraženiji u odnosu na postupan razvoj simptoma i znakova kod PACNS-a. Lumbalna punkcija kod bolesnika s RCVS-om obično je uredna, a MR mozga ne pokazuje zone ishemije, a na MR angiografiji može se naći znak krunice kao i kod PACNS-a. Navedene promjene kod ovih bolesnika su reverzivilne te se povlače unutar jednoga do dva mjeseca (uredni nalaz kontrolne MR angiografije jedan je od najsigurnijih nalaza u postavljanju ove dijagnoze). Osnovu liječenje RCVS-a čini primjena kalcijskih blokatora, a u nejasnim situacijama i teškom razlučivanju od PACNS-a u terapiju se uvode i glukokortikoidi. Prognoza ovih bolesnika je povoljna.
SERONEGATIVNI SPONDILOARTRITISI
Uvod. Seronegativni spondiloartritisi predstavljaju skupinu upalnih reumatskih bolesti koje karakteriziraju određena skupna obilježja kao što su asimetrično zahvaćanje perifernih zglobova i aksijalnoga skeleta (sakroilijakalni zglobovi i kralješnica), genetska predispozicija (više od 90 % oboljelih ima HLA-B27 antigen) te odsutnost reumatoidnoga faktora. Osim koštano-mišićnih struktura, ovim bolestima mogu biti zahvaćena i druga tkiva i organi, kao što su koža, oči, srčani zalisci i aorta. Procijenjena prevalencija seronegativnih spondiloartritisa u Europi kreće se između 1 i 2 %. U ovu skupinu bolesti ubrajamo ankilozantni spondilitis, psorijatični artritis, reaktivni artritis, enteropatski artritis te nediferencirani artritis.
ANKILOZANTNI SPONDILITIS (Morbus Bechterew)
Definicija. Ankilozantni spondilits (AS) predstavlja seronegativni spondiloartritis kojeg karakterizira zahvaćanje aksijalnoga skeleta, posebice sakroilijakalnih zglobova i velikih proksimalnih zglobova. Počinje nespecifičnim upalnim promjenama zglobnih sveza (entezitis), a završava stvaranjem nove kosti, okoštavanjem i razvojem progresivne ankiloze (gubitak zglobne pokretljivosti).
Epidemiologija. Procijenjena prevalencija AS-a u općoj populaciji iznosi oko 0,2 %, dok se u populaciji HLA-B27 pozitivnih osoba ona kreće oko 2 %. AS se češće javlja kod osoba muškoga spola s omjerom od 2,5:1 do 5:1, i to obično u drugom i trećem desetljeću života, te pokazuje teži klinički tijek nego kod žena. Kod 15 do 20 % oboljelih osoba bolest nalazimo i kod najbližih srodnika što upućuje na snažnu genetsku poveznicu i uporište.
Etiopatogeneza. Vjeruje se da AS nastaje kao posljedica poremećenoga imunološkog odgovora koji nastaje kod genetski predisponiranih pojedinaca, a potaknut je nekim vanjskim čimbenikom. Na genetsku predispoziciju ukazuje povećana učestalost bolesti kod osoba koje imaju HLA-B27 antigen, ali danas se zna da i drugi geni mogu imati određenu ulogu u patogenezi AS-a, ponajprije geni koji sudjeluju u unutarstaničnim signalnim putevima upalnih medijatora TNF (TNFRSF1A, LTBR, TBKBP1) te IL-23 i IL-17 (IL23R, PTER4, IL12B, CARD9, TYK2) koji imaju ključnu ulogu u nastanku AS-a. Do sada nije utvrđen točan egzogeni pokretač cijeloga patološkog procesa, ali sve se više govori o utjecaju crijevnih bakterija i mikrooštećenju crijevne sluznice genetski predisponiranih pojedinaca. Pretpostavljeni mehanizam podrazumijeva penetraciju komponenata bakterijske stijenke (antigeni) kroz oštećenu mukoznu barijeru u submukozu gdje dovode do aktivacije makrofaga i dendritičkih stanica koje potom luče IL-23 koji dalje aktivira T-limfocite, neutrofile i mastocite koji potom luče IL-17, IL-22 i TNF-α koji su odgovorni za nastanak ranih upalnih promjena u sakrolijakalnim i drugim zglobovima. Nije poznato zašto pokrenuti mehanizam i navedeni medijatori i T-limfociti pokazuju organotropiju prema zglobnim svezama aksijalnoga skeleta. Smatra se da IL-23/IL-17 osovina ima ključnu ulogu u ovim događajima. Rane promjene uključuju upalu zglobnih sveza (entezitis). Kasnije u tijeku bolesti vidljivi su i rezultati učinka TNF-α te ostalih proupalnih citokina, dovođenjem do redukcije razine Dickkopf-1 i sklerostina što za posljedicu ima formiranje nove kosti, što je jedna od glavnih obilježja ove bolesti, a dovodi do ankiloze i gubitka funkcije zgloba. Kako se pretpostavlja da je pokretački antigen zapravo dio bakterije koju nalazimo u sastavu fiziološke crijevne flore, sve se više o AS-u govori kao autoupalnoj, a manje kao autoimunoj bolesti.
Patologija. AS prvom redu zahvaća zglobove i spojeve aksijalnoga skeleta (sakroilijakalni zglobovi, intervertebralni diskovi i kostovertebralni zglobovi), nešto rjeđe zahvaća velike proksimalne zglobove (kukovi, koljena, ramena), a gotovo nikada ne zahvaća male zglobove šaka i stopala. Upalni infiltrat u AS-u u prvom se redu sastoji od T-limfocita (CD4+, CD8+), a nalaze se i neutrofili, makrofazi i u manjoj mjeri B-limfociti. U kasnijim fazama bolesti sinovitis prelazi u fibrozirajući proces nakon kojega slijedi okoštavanje i ankiloza.
Klinička slika. AS karakterizira spor i progresivan klinički tijek. Najraniji simptomi uključuju bol u donjem dijelu leđa koji se spušta prema glutealnoj regiji, a najizraženiji je u jutarnjim satima nakon buđenja i povezan je s ukočenošću donjega dijela leđa koja traje satima. Za razliku od degenerativnih promjena (spondiloza), kod bolesnika s AS-om bolovi i ukočenost prolaze i poboljšavaju se s razgibavanjem i tjelesnom aktivnosti. S napredovanjem bolesti bol i ukočenost se šire prema kranijalno i zahvaćaju sve veći dio kralješnice, a pokreti postaju bolni i otežani. U razvijenoj fazi bolesti kada dolazi do nastanka ankiloze bolesnici razvijaju specifične deformitete kralješnice i kukova (izražena zakrivljenost prema naprijed koju uvjetuje torakalna kifoza i gubitak fiziološke lumbalne lordoze, fleksijske kontrakture kukova). Ostale kliničke manifestacije AS-a uključuju prolazni periferni artritis, entezitis Ahilove tetive, prednji uveitis, aortritis, smetnje interventrikularnoga provođenja, plućnu fibrozu, amiloidozu te IgA nefropatiju.
Dijagnoza AS-a postavlja se na temelju kliničke slike i kliničkoga nalaza, laboratorijskih nalaza te slikovnih metoda. Bolnost i ukočenost u donjem dijelu leđa, posebice kod mladih muškaraca, mora pobuditi sumnju na AS. Laboratorijski nalazi ukazuju na ubrzanu sedimentaciju eritrocita i umjerenu anemiju kronične bolesti. Bolesnici imaju negativan reumatoidni faktor i anticitrulinska antitijela, a kod većine dokazujemo pozitivan HLA-B27 antigen (> 90 %). Početne promjene na sakroilijakalnim zglobovima u smislu entezitisa moguće je dokazati samo MR snimanjem kralježnice (magnetna rezonanca), dok se u kasnijim fazama, kada dolazi do sklerozacije i ankiloze, promjene mogu detektirati i klasičnim radiološkim snimanjem (u uznapredovaloj fazi okoštala kralježnice ima "znak bambusovoga štapa"). Ako su promjene na zglobovima vidljive na klasičnom rendgenogramu, tada govorimo o ankilozantnom spondilitisu, a ako se radi o ranoj bolesti koja nema još uočljive promjene na klasičnom rendgenogramu, već su one vidljive na magnetskoj rezonaciji sakroilijakalnih zglobova, tada govorimo o neradiografskom seronegativnom spondilitisu (nrSpA).
Dijagnostički kriteriji. U dijagnostici AS-a koristimo se ASAS kriterijima (engl. The Assessment of Spondyloarthritis International Society) iz 2009. godine. Osnovni preduvjet (ulazni kriterij) za postavljanje dijagnoze prema ovim smjernicama podrazumijeva postojanje bolova u leđima u trajanju dužem od tri mjeseca kod osoba mlađih od 45 godina. Ako je zadovoljen taj ulazni kriterij, daljnje se bodovanje vrši prema prisutnosti drugih kriterija koji su svrstani u dvije skupine: primarni i sekundarni (Tablica 9.19). Dijagnoza se postavlja ako nalazimo znakove sakroileitisa na MR-u (ili klasičnom rendgenogramu) uz minimalno jedan (ili više) sekundarni kriteriji ili HLA-B27 pozitivni antigen uz minimalno dva (ili više) sekundarnih kriterija.
|
Tablica 9.19. ASAS kriteriji za aksijalni spondiloartritis
|
|
Ulazni kriterij
|
Bol u leđima u trajanju > 3 mjeseca u dobi < 45 godina
|
|
Primarni kriteriji
|
Dokaz sakroileitisa (MRI, RTG), pozitivan HLA-B27 antigen
|
|
Sekundarni kriteriji
|
Bol u leđima, artritis, entezitis, uveitis, daktilitis, psorijaza, Crohnova bolest, dobar odgovor na NSAID, obiteljska anamneza AS-a, povišen CRP
|
Liječenje. Prvu i osnovnu liniju liječenja čini primjena nesteroidnih protuupalnih lijekova čime se kod velike većine bolesnika postiže odgovarajuća kontrola bolesti. Osim što smanjuju simptome, oni sprečavaju napredovanje same bolesti i preveniraju nastanak ankiloze. Zajedno s primjenom NSAR-a, bolesnici se podvrgavaju i mjerama fizikalnoga liječenja i rehabilitacije. Kod bolesnika koji ne reagiraju na NSAR-e danas se preporuča liječenje biološkim lijekovima, u prvom redu TNF inhibitorima, kao što su etanercept (50 mg sc jednom tjedno), adalimumab (40 mg sc svaki drugi tjedan), infliksimab (5 mg/kg iv svaki drugi mjesec), golimumab (50 mg sc jednom mjesečno) ili certolizumab (200 mg sc svaki drugi tjedan). U obzir dolazi i primjena IL-17 inhibitora (sekukinumab, iksekizumab) te IL-12/IL-23 inhibitora (ustekinumab). Glukokortikoidi i drugi imunosupresivi i imunomodulatori nemaju veći učinak u liječenju ovih bolesnika, pa se ne koriste u kliničkoj praksi.
Prognoza. Konačni ishod ovih bolesnika uvelike je promijenila era bioloških lijekova, s obzirom na progresivan tijek bolest, osobito kod bolesnika koji ne reagiraju na liječenje NSAR-om i koji bi završavali s anikolozom, deformitetima kralježnice i velikim invalidtetom. Uz današnje mogućnosti liječenja kod gotovo svih bolesnika postiže se simptomatsko olakšanje, usporenje progresije bolesti te očuvanje kvalitete života i izbjegavanje trajne invalidnosti.
PSORIJATIČNI ARTRITIS
Definicija. Psorijatični artritis (PsA) predstavlja seronegativni spondiloartritis koji karakterizira kronična upala zglobova koja se javlja kod osoba oboljelih od psorijaze, kronične kožne bolesti obilježene proliferacijom keratinocita, pojačanim ljuskanjem i crvenilom. Ona se najčešće prezentira erozivnim oligo ili poliartririsom, daktilitisom i nešto rjeđe, entezitisom sakroilijakalnih zglobova i aksijalnoga skeleta.
Epidemiologija. Procijenjena prevalencija PsA u općoj populaciji kreće se od 1 do 3 %, dok se u populaciji osoba koje boluju od psorijaze ona kreće oko 30 do 40 %. Bolest se najčešće manifestira u trećem i četvrtom desetljeću života, podjednako u oba spola.
Etiopatogeneza. Potpuno poznavanje etiopatogeneze PsA nije poznato, a smatra se da nastaje kompleksnom interakcijom genetskih i okolišnih čimbenika koji rezultiraju poremećenim imunoodgovorom usmjerenim na zglobove i druga tkiva. Na genetsku osnovu bolesti ukazuje povećana pojavnost unutar nekih obitelji i između bližih srodnika, snažna povezanost s od ranije poznatim HLA haplotipovima (HLA B13, B17, B19, B37, B39, Cw6.) te novije otkrivenim genima koji sudjeluju u procesima unutarstanične signalizacije IL-12 i IL-23. Od okolišnih čimbenika kao pokretača bolesti, PsA najčešće se povezuje s mehaničkom traumom te virusnim infekcijama, a kao mogući čimbenici spominju se i pušenje, imunizacija i stres. Pretpostavljeni mehanizam podrazumijeva aktivaciju dendritičkih stanica u području kože i sluznica potaknutu nekim vanjskim pokretačem (antigenom), što dovodi do stvaranja i aktivacije specifičnih CD8+ T-limfocita koji se smatraju ključnima u nastanku PsA. Aktivirani T-limfociti vežu se na ciljne antigene te posredovanjem transkripcijskih faktora i aktivacijskih proteina pokreću upalnu reakciju koja rezultira konačnim oštećenjem tkiva i organa (koža, sinovijalna membrana, enteze, zglobna čahura). Novija istraživanja sugeriraju i na ulogu CD4+ T-limfocita čiji su aktivator također dentritičke stanice putem IL-23, osobito subpopulacije Th17 koji dominantno stvaraju IL-17 koji se u povećanoj mjeri nalazi u sinovijalnoj tekućini i hrskavičnom tkivu oboljelih osoba, zbog čega se i smatra da ima veliku ulogu u posredovanju nastalih promjena i oštećenja.
Klinička slika. Kod većine bolesnika (60 - 70 %) psorijatične kožne promjene prethode pojavi PsA, dok kod manjega broja bolesnika (15 - 20 %) nalazimo zahvaćenost zglobova prije nastupa kožnih promjena. Kod svega manje od 5 % oboljelih kožne i zglobne promjene javljaju se unutar iste godine. Kliničke manifestacije PsA mogu uključivati zahvaćanje perifernih zglobova ili zglobova aksijalnoga skeleta, a moguće su i druge pridružene kliničke manifestacije (Slika 9.7.).
Periferna bolest. Zahvaćanje perifernih zgloba temeljno je obilježje PsA, a može se prezentirati na jedan od sljedećih načina: klasični oblik sa zahvaćenim noktima i distalnim interfalanegalnim zglobovima, mutilirajući artritis s osteolizom falangi, simetrični poliartritis sličan reumatoidnom artritisu ili asimetrični mono ili oligoartritis s često pridruženim daktilitisom (difuzna upala cijeloga prsta). Zahvaćeni zglobovi pokazuju tipična obilježja upale: otok, crvenilo, toplina, bol i poremećena funkcija (pokretljivost).
Aksijalna bolest. Kod više od 50 % bolesnika s PsA možemo naći i znakove aksijalnoga artritisa (spondilitis, sakroiliitis). Ona je najčešće udružena s perifernim artritisom i rijetko se pojavljuje u samostalno. Kod tih bolesnika nalazimo tipičnu upalnu bolnost kralježnice, osobito donjega dijela, koja se pogoršava u mirovanju, budi bolesnike tijekom noći te se smiruje pri tjelesnim kretnjama i aktivnostima te dobro reagira na NSAR-e. Kod nekih bolesnika prisutna je i ograničena pokretljivost kralježnice. Progresija prema ankilozi za razliku od bolesnika s AS-om ovdje se rjeđe viđa i nije karakteristična za PsA.
Ostale manifestacije. Većina bolesnika navodi i bolnost enteza (hvatišta tetiva, ligamenata, fascija i zglobnih čahura), a najčešće se radi o Ahilovoj tetivi i plantarnoj fasciji. Daktilitis označava difuznu upalu cijeloga prsta, a karakteriziran je kobasičastom, bolnom i crvenom oteklinom (najčešće asimetrično zahvaća po nekoliko prstiju ruku i nogu). Uz navedene manifestacije bolesnici mogu imati i izražene psorijatične promjene te promjene na drugim tkivima i organima (oči, probavni sustav, srčano-žilni sustav te mokraćni sustav).
Slika 9.7. Psorijaza ruku i noktiju (lijevo) i mutilirajući oblik psorijatičnoga artritisa (desno)
Dijagnoza se postavlja na temelju kliničke slike i fizikalnoga pregleda, laboratorijskih nalaza te slikovnih metoda. Pri postavljanju dijagnoze bitni su anamnestički podaci o prisutnosti kožnih promjena, kao i pojavnosti PsA kod bližih srodnika. U laboratorijskim nalazima možemo naći umjerenu anemiju kronične bolesti, povišene vrijednosti C-reaktivnoga proteina i sedimentacije eritrocita te hiperuricemiju kao posljedicu pojačane izmjene kožnih stanica. Većina bolesnika ima pozitivan HLA-B27 antigen te imaju negativan reumatoidni faktor i anticitrulinska antitijela, iako to nije glavni kriterij budući da oni mogu biti pozitivni kod oko 5 % ovih bolesika. Slikovne metode služe za objektivizaciju zglobnih promjena, a pri tome se koristimo klasičnim radiografskim snimanjem (periferni zglobovi) ili magnetnom rezonancom (aksijalni skelet). U početnim fazama bolesti klasičan radiografski nalaz bit će uredan. U dijagnostici entezitisa koristi se ultrazvučnim pregledom.
Dijagnostički kriteriji. U postavljanju dijagnoze PsA koriste se CASPAR kriterijima (izv. Clasiffication of psoriatic arthritis). Dijagnoza se postavlja ako uz upalnu bolest zgloba, kralježnice ili enteza bolesnik ima barem tri boda iz navedenih pet kategorija (Tablica 9.20.).
|
Tablica 9.20. CASPAR kriteriji za postavljanje dijagnoze PsA
|
|
Podaci o psorijazi
|
Trenutno prisutne psorijatične promjene
|
2 boda
|
|
Pozitivna osobna anamneza na psorijazu
|
1 bod
|
|
Pozitivna obiteljska anamneza na psorijazu
|
1 bod
|
|
Psorijatične promjene noktiju
|
1 bod
|
|
Nalaz negativnoga reumatoidnog faktora
|
1 bod
|
|
Daktilis prisutan u trenu pregleda ili u osobnoj anamnezi
|
1 bod
|
|
Jukstaartikularno stvaranje nove kosti (radiološki potvrđeno)
|
1 bod
|
Liječenje. Prvu liniju liječenja PsA čini primjena NSAR-a koji su dovoljni u kontroli simptoma i progresije bolesti kod bolesnika s blažim sinovitisom i aksijalnim zahvaćanjem. Kod bolesnika s perifernim artritisom NSAR treba kombinirati s konvencionalnim sintetskim DMARD (csDMARD) lijekovima, u prvom redu metotreksatom, s obzirom na njegovu učinkovitost i na kožne i na zglobne manifestacije bolesti (makismalna doza metotreksata je 25 mg jednom tjedno), a u drugom redu dolaze u obzir i leflunomid (doza 20 mg dnevno) te sulfasalazin (do 3 gr dnevno). Kod bolesnika koji su rezistentni i na primjenu metotreksata (ili drugih csDMARD) drugu liniju liječenja čini primjena biološke DMARD (bDMARD) terapije: TNF inhibitora (vrste i doze iste su kao i kod AS), inhibitorima IL-12/IL-23 (ustekinumab) ili IL-17 (sekukinumab, iksekizumab) te inhibitorima Il-17/IL-23 (risenkizumab). Ako se ne postiže zadovoljavajući odgovor na kombinaciju csDMAR i bDMARD terapije, ili je bDMARD terapija kontraindicirana, postizanje kontrole bolesti može se pokušati s JAK inhibitorima (tofacitinib). Glukokortikoidi ne pokazuju značajniji učinak kod ovih bolesnika pa se ne koriste u klasičnom liječenju, a njihova uporaba može pogoršavati pojavu eritrodermije u psorijazi. Kod bolesnika s blažim oblikom bolesti koji ne reagiraju na konvencionalne sintetske DMARD lijekove, te kod onih koji ne žele biološku terapiju ili je ona kontraindicirana, može se pokušati i s primjenom apremilasta, inhibitora fosfodiesteraze 4 koji ima određena imunomodulatorna djelovanja.
Prognoza. Uz pravovremeno postavljanje dijagnoze i liječenje većina bolesnika ima dobru prognozu i postiže se zadovoljavajuća kontrola bolesti. Manje od 5 % bolesnika ima težak oblik bolesti sa stvaranjem deformiteta i gubitkom funkcije zahvaćenih zglobova.
REAKTIVNI ARTRITIS
Definicija. Reaktivni artritis (ReA) predstavlja aseptičnu (sterilnu) upalu zglobova koja nastaje kao rezultat imunološki posredovanoga odgovora na nedavne gastrointestinalne ili urogenitalne infekcije tako da razlikujemo postenterički i postvenerički ReA. S obzirom na njegove karakteristike, kao što su periferni asimetrični monoartritis ili oligoartritis te povezanost s HLA-B27 antigenom, ReA se ubraja u skupinu seronegativnih spondiloartritisa.
Epidemiologija. Procijenjena godišnja incidencija ReA različita je te se kreće između 1 i 25 na 100 000 stanovnika. ReA će se javiti kod 1 do 3 % osoba nakon preboljele genitourinarne infekcije, odnosno kod 1 do 4 % osoba nakon gastrointestinalne infekcije kada se promatra opća populacija, dok se taj postotak znatno povećava za HLA-B27 pozitivne osobe i iznosi 30 do 40 %. ReA se javlja najčešće u srednjoj životnoj dobi (drugo do četvrto desetljeće života) s većom učestalošću kod muškaraca u odnosu na žene kada se radi o postveneričnom obliku (5 - 10:1), dok je taj odnos ravnomjeran kada se radi o postenteričkom obliku (1:1).
Etiopatogeneza. Pretpostavljeni mehanizam nastanka ReA uključuje nekoliko patofizioloških rekacija koje se zbivaju u zahvaćenom zglobu, a povezane su s preboljenim infekcijama (bakterije koje se povezuju s razvojem ReA navedene su u Tablici 9.21.). S obzirom na to da se u zahvaćenim zglobovima gotovo svih bolesnika mogu identificirati bakterijski antigeni, jedna od osnovnih pretpostavki je da su oni odgovorni za nastalo upalno zbivanje, iako nije razjašnjena njihova sklonost da se odlažu i nakupljaju u sinovijalnoj membrani. Tom procesu mogu pridonijeti i HLA-B27 antigeni koji pokazuju određenu sličnost s bakterijskim antigenima što se objašnjava molekularnom mimikrijom i reakcijom križne reaktivnosti, a dijelom objašnjava i teže kliničke oblike i češće recidive ReA kod tih bolesnika.
|
Tablica 9.21. Infektivni uzročnici povezani s ReA
|
|
Gastrointestinalne infekcije
|
Urogenitalne infekcije
|
|
Salmonella spp
Shigela spp
Yersinia spp
Campylobacter spp
Clostridium difficile
Escherichia coli
|
Chlamydia trachomatis
Mycoplasma genitalium
Ureaplasma urealyticum
|
Klinička slika. Klasično, ReA se javlja 1 do 4 tjedna nakon simptomatske ili asimptomatske urogenitalne ili gastrointestinalne infekcije. Početak bolesti je obično akutan (rjeđe subakutan), a manifestira se općim, zglobnim i izvanzglobnim manifestacijama. Opće (sistemske) manifestacije podrazumijevaju umor i malaksalost, gubitak teka te povišenu tjelesnu temperaturu.
Zglobne manifestacije. ReA obično se očituje kao asimetrični oligoartritis ili monoartritis koji zahvaća velike zglobove pretežno donjih ekstremiteta sa zahvaćanjem periartikularnih struktura (tendinitis, tendosinovitis, peritendinitis). Zahvaćeni zglobovi su otečeni, bolni i crveni s poremećenom pokretljivošću. Zahvaćenost malih zglobova šaka i stopala nisu česta pojava u ReA, kao niti unilatrealni ili bilateralni sakroileitis (upalna križobolja) te se oni mogu naći kod oko trećine bolesnika. Kod oko 10 % bolesnika mogu se naći kobasičasto otečeni pojedini prsti šaka ili stopala (daktilitis). Zglobne promjene mogu biti migratorne prirode (zahvaćenost zgloba seli se s jednoga na drugi zglob) te mogu trajati u manjem ili jačem intenzitetu i do godine dana.
Izvanzglobne manifestacije. Mukokutane (kožne) i okularne (očne) manifestacije najčešće su izvanzglobne manifestacije kod ovih bolesnika. Trijas koji čini artritis, konjuktivitis i uretritis naziva se Reiterov sindrom. Mukokutane manifestacije uključuju pojavu pustuloznoga palmo-plantarnog egzantema, bezbolnih oralnih ulceracija te promjena na koži prepucija (balanitis, ulceracije). Okularne manifestacije mogu se očitovati kao konjuktivitis, iritis, prednji uveitis, episkleritis ili retrobulbarni neuritis. Ostale izvanzglobne promjene javljaju se izuzetno rijetko, a uključuju afekciju miokarda (miokarditis, poremećaji provođenja), perifernoga i središnjega živčanog sustava s posljedičnim neurološkim ispadima te pluća (pleuritis, pneumonitis) i bubrega (proteinurija).
Dijagnoza se postavlja na osnovi kliničke slike, anamnestičkih (epidemioloških) podataka, laboratorijskih nalaza i slikovnih radioloških metoda. Ključan podatak u postavljanju dijagnoze ReA je preboljena infekcija probavnoga ili genitourinarnoga sustava, iako one često prolaze asimptomatski, osobito spolno prenosive infekcije. U akutnoj fazi u laboratorijskim nalazima bilježimo leukocitozu, ubrzanu sedimentaciju eritrocita te povišeni C-reaktivni protein. U široj laboratorijskoj obradi određujemo HLA-B27 koji je pozitivan kod više od 80 % oboljelih te reumatoidni faktor i anticitrulinska antitijela u diferencijalno dijagnostičke svrhe, s obzirom na to da su kod ovih bolesnika negativni. Kod svih bolesnika sa sumnjom na ReA potrebno je provesti mikrobiološku dijagnostiku koja podrazumijeva dijagnostiku genitalnoga sustava na CUM infekcije (Chlamydia, Ureaplasma, Mycoplasma) što se radi uzimanjem brisa cerviksa ili uretre ili određivanjem antigena iz urina te, prema indikaciji, mikrobiološku obradu stolice. Klasični rendgenogram u akutnoj fazi bolesti najčešće je uredan, dok u kroničnoj fazi možemo naći znakove erozivnoga artritisa. Zahvaćenost mekotkivnih periartikularnih struktura najbolje detektiramo ultrazvučnim pregledom.
Liječenje. Osnovu liječenja čini primjena antibiotika s ciljem eradikacije infekta, a zglobne i izvanzglobne manifestacije u prvoj liniji liječimo primjenom NSAR-a u kombinaciji s glukokortikoidima u težim oblicima (metilprednizolon iv ili prednizon per os ili intraartikularna primjena depo preparata). U slučaju učestalih relapsa ili nemogućnosti postizanja kontrole bolesti s NSAR-om i glukokortikoidima, u drugoj liniji liječenja primjenjuje se sulfasalazin (2 x 1 g/dan) ili metotreksat (7,5 - 20 mg/tjedno), dok kod bolesnika refraktornih na liječenje NSAR-ima i csDMARD lijekovima u obzir dolazi primjena TNF inhibitora, kao i kod ostalih seronegativnih spondiloartritisa.
Prognoza je uz pravilno liječenja povoljna kod svih bolesnika. Kod jednoga dijela bolesnika, osobito onih s nesaniranim žarišnim infektom mogući su učestali recidivi, odnosno egzacerbacije simptoma.
ENTEROPATSKI (SPONDILO)ARTRITIS
Definicija i epidemiologija. Enteropatski spondiloartritis podrazumijeva pojavu perifernoga ili aksijalnog artritisa kod bolesnika s idiopatskom upalnom bolesti crijeva (Crohnova bolest, ulcerozni kolitis), a predstavlja jednu od najčešćih ekstraintestinalnih manifestacija ovih poremećaja koja se može naći kod 5 do 20 % tih bolesnika, i to češće kod bolesnika koji boluju od Crohnove bolesti.
Etiopatogeneza. Točan uzrok i mehanizam nastanka ovoga oblika artritisa nije poznat. Smatra se da ključnu ulogu ima upala crijevne stijenke koja omogućava lakši prodor intraluminalnih antigena u submukozu i krvotok, koji dalje dovode do aktivacije imunološkoga sustava. Upala sinovijalne membrane može biti posljedica odlaganja samih antigena u nju i aktivacijom imunološkoga sustava u samom zglobu ili je posljedica molekularne mimikrije i križne reakcije između antigena HLA-B27 i izvršnih stanica imunološkoga sustava aktiviranih u crijevnoj stijenci.
Klinička slika. Enteropatski arteritis povezan s upalnom bolesti crijeva generalno se može očitovati kao aksijalni artritis (sakroileitis i/ili spondilitis) te kao periferni artritis.
Glavne kliničke karakteristike aksijalnoga artritisa povezanoga s upalnom bolesti crijeva su: pojava nagle boli u donjem dijelu leđa, jutarnja ukočenost, pogoršanje kod dugotrajnoga stajanja ili sjedenja te poboljšanje kod umjerene aktivnosti. Pojava ovih simptoma nije povezana s intenzitetom gastrointestinalnih simptoma.
Periferni artritis kod bolesnika s upalnom bolesti crijeva po svojim karakteristikama je neerozivan i ne dovodi do trajnih oštećenja ili deformiteta. Periferni artritis može se pojaviti zajedno s intestinalnim simptomima, ali se može dogoditi i da se zglobni simptomi jave prije intestinalnih. Razlikujemo dva klinička oblika: tip I i tip II.
Tip I zahvaća manje od pet zglobova, a karakteristično je asimetrično zahvaćanje velikih zglobova (koljeno, kuk, rame). Javlja se naglo, brzo se razvija i ne traje duže od 10 tjedana. Obično prethodi intestinalnim simptomima, ali se može pojaviti i u vrijeme egzacerbacije intestinalnih simptoma. Povezan je i s drugim ekstraintestinalnim manifestacijama crijevne upalne bolesti.
Tip II zahvaća više od pet zglobova, a karakteristično je simetrično zahvaćanje malih zglobova šaka i stopala (nalikuje reumatoidnom artritisu). Daleko se rjeđe javlja od tipa I. Radi se o promjenama koje se sporo razvijaju, kroničnoga karaktera, a traju mjesecima, nekada i do godine dana.
Dijagnoza se postavlja na temelju kliničke slike, anamnestičkih podataka (podatak o idiopatskoj crijevnoj upalnoj bolesti), laboratorijskih nalaza te slikovnih metoda. Laboratorijski nalazi ponajprije ovise o intenzitetu crijevne bolesti, ali se načelno bilježi ubrzana sedimentacija eritrocita, leukocitoza te povišeni upalni markeri. Kod većine bolesnika nalazimo pozitivan HLA-B27 antigen. Slikovne radiološke metode ovise o jačini upalnoga procesa i izraženosti oštećenja, ali s obzirom na to da se radi o neerozivnim artritisima, nalazi su uglavnom uredni.
Liječenje. Osnovu liječenja čini liječenje idiopatske upalne bolesti crijeva kao etiološkoga čimbenika, a primjena nesteroidnih upalnih lijekova s ciljem poboljšanja zglobnih simptoma mora biti s oprezom, budući da oni mogu pogoršati upalnu bolest crijeva. Glukokortikoidi se koriste za kratkotrajno liječenje kod egzacerbacija artikularnih simptoma, kada se kontrola ne može postići primjenom NSAID-a, a mogu se primjenjivati enteralno, parenteralno i lokalno (infiltracija zgloba). Kod bolesnika s izraženim intestinalnim i ekstraintestinalnim simptomima koji ne reagiraju na klasične metode liječenja upalne bolesti crijeva, opravdano je liječenje biološkom terapijom, odnosno TNF inhibitorima (infikimab, adalimumab), Il.12/IL-23 inhibitorima, te JAK inhibitorima
Prognoza. Tijek i težina enteropatskih artritisa je varijabilna. Prognoza ponajprije ovisi o težini crijevne bolesti koja je i dovela do artritisa. Prosječno je potrebno oko šest tjedana da dođe do povlačenja simptoma bolesti nakon njihovoga pojavljivanja. Same zglobne promjene imaju dobru prognozu te se najčešće smiruju i prolaze bez trajnih posljedica.
NEDIFERENCIRANI SPONDILOARTRITIS
Nediferencirani spondilartritis je naziv za seronegativne spondiloartritise koje ne možemo svrstati niti u jednu od navedenih skupina. Često se koristi kao radna dijagnoza, a većina pacijenata kroz određeno vrijeme razvije sliku nekoga od specifičnih oblika artritisa. Liječenje je individualno.
RELAPSIRAJUĆI POLIHONDRITIS
Definicija. Relapsirajući polihondritis (RP) predstavlja imunološki posredovanu bolest obilježenu recidivirajućim upalama koje zahvaćaju hrskavična i proteoglikanima bogata tkiva, najčešće uha, nosa, perifernih zgloba te traheobronhalnoga stabla dovodeći do anatomskih deformiteta i funkcionalnih ispada.
Epidemiologija. Radi se o rijetkom poremećaju čija se incidencija procjenjuje na oko 3,5 slučaja na milijun stanovnika godišnje. Iako se RP može pojaviti u bilo kojoj životnoj dobi, najčešće se javlja u četvrtom i petom desetljeću života, podjednako kod oba spola. Kod 30 % bolesnika s RP-om nalazimo i druge imunološki posredovane bolesti, najčešće reumatoidni artritis.
Etiopatogeneza. Točan uzrok i mehanizam razvoja RP-a nije poznat. Kao i kod drugih sličnih bolesti, uočena je povećana pojavnost bolesti kod osoba koje imaju HLA-DR4 antigene. Smatra se da se u podlozi radi o poremećenom imunološkom (humoralnom i staničnom) odgovoru na određene antigene unutar hrskavičnoga tkiva čiji se pokretački događaj ne zna (infekcija ili mehaničko oštećenje), a u konačnici rezultira destrukcijom hrskavičnoga tkiva. U prilog tome da se radi o poremećenom humoralnom imunoodgovoru govori nalaz pozitivnih autoantitijela protiv kolagena tipa II, kao i nekih drugih hrskavičnih antigena (matrilin-1, oligomerični proteini matriksa), dok na poremećaj staničnoga imunoodgovora upućuje patohistološki nalaz upalnoga infiltrata kojeg u najvećoj mjeri čine T-limfociti, osobito CD4+. Tijekom ovih zbivanja u pojačanoj mjeri stvaraju se i otpuštaju proteolitički enzimi koji dovode do hrskavičnih oštećenja s nastankom deformiteta i poremećenom funkcijom.
Patologija. Inicijalne lezije u RP-u su hondritis i poliartritis koji se mogu javiti na različitim mjestima što može stvarati raznoliku kliničku sliku i time otežati postavljanje prave dijagnoze. Također, osim zahvaćanja hrskavičnih struktura, slične lezije mogu se naći i na anatomskim strukturama bogatim proteoglikanima kakvi su oči, srčani zalisci i krvne žile. Patohistološki, bolest je u početnim fazama obilježena edemom i upalnim infiltratom kojega čine T-limfociti (dominantno), makrofagi, plazma stanice i depoziti imunokompleksa, dok se u kasnijim fazama jasno uočavaju lezije i strukturna oštećenja hrskavičnoga tkiva.
Klinička slika. Bolest karakterizira epizodna pojavnost sa zahvaćanjem različitih hrskavičnih i tkiva bogatih proteoglikanima. Najkarakterističniji klinički nalaz uključuje hondritis usne školjke, nosnih hrskavica te laringotraheobronhalnih hrskavica. Ostali klinički nalazi uključuju artropatiju, očne, neurološke, renalne, dermatološke i kardiovaskularne manifestacije.
Hondritis. Jednostrana ili obostrana upala ušnih hrskavica temeljna je klinička minifestacija RP-a i javlja se kod oko 90 % bolesnika. Nastaje naglo, a obilježeno je crvenilom, edemom i palpatornom bolnošću, a ima tendenciju spontanoga prolaska te ponovnoga javljanja u različitim vremenskim intervalima. Postupno dolazi do razvoja deformiteta ušne školjke, a kod manjega broja bolesnika moguć je i razvoj provodnoga gubitka sluha uslijed kolapsa aurikularne hrskavice i edema slušnoga kanala. Upala nosne hrskavice javlja se kod manje od 50 % bolesnika s RP-om, a obilježena je demom, crvenilom i bolnošću korijena nosa uz povremene epistakse. U kroničnoj formi razvija se sedlasta malformacija nosa. Zahvaćenost hrskavica laringotraheobronhalnoga stabla javlja se kod oko 10 % bolesnika, ali predstavlja najozbiljnije kliničko očitovanje uslijed kompromitiranja dišnoga puta, u akutnoj fazi edemom, a u kroničnoj fazi strikturama i deformacijama.
Artropatija. Zahvaćanje zglobova u RP-u nalazimo u 50 do 80 % bolesnika, a obično se razvija nakon hondritisa (vrlo rijetko se javlja kao inicijalna klinička manifestacija). Najčešće se radi o asimetričnom intermitentnom oligoartritisu ili poliartritisu koji pogađa metakarpofalanegalne i proksimalne interfalangealne zglobove, a rjeđe laktove, koljena, gležnjeve i metatarzofalangealne zglobove stopala. Zahvaćenost aksijalnoga skeleta nije karakteristična za RP.
Ostale manifestacije. Okularne manifestacije nalazimo kod oko 50 do 60 % bolesnika, najčešće u vidu skleritisa i episkleritisa te konjuktivitisa dok su iritis, retinopatija, optički neuritis, periferni keratitis i retinalni vaskulitis rijetki. Ostale kliničke manifestacije su rijetke i javljaju se kod manje od 20 % bolesnika, a uključuju neurološke manifestacije (meningitis, encefalitis, demencija, psihoza), renalne manifestacije (IgA nefropatija, tubulointersticijski nefritis, membranska nefropatija), dermatološke manifestacije (purpura, livedo reticularis, distalne ulceracije i nekroze) te kardiovaskularne manifestacije (valvularna bolest srca, aneurizme i disekcija aorte, miokarditis, perikarditis, poremećaji provođenja).
Dijagnoza. Postavljanje dijagnoze RP-a predstavlja klinički izazov s obzirom na širok dijapazon kliničkih manifestacija. Temeljno i ishodišno polazište u procesu dijagnostike predstavlja klinička slika u kojoj dominiraju opisani hondritis i artritis, a na to se nadovezuju laboratorijske i slikovne metode. Neki od predloženih dijagnostičkih kriterija za RP prikazani su u Tablici 9.22.
Laboratorijske metode. U osnovnim laboratorijskim nalazima mogu se naći anemija kronične bolesti te povišeni upalni markeri (C-reaktivni protein, sedimentacija eritrocita) u fazi klinički manifestne bolesti. Kod trećine bolesnika mogu se naći poliklonalna hipergamaglobulinemija te pozitivna antinuklearna antitijela (ANA) i antineutrofilna citoplazmatska antitijela (ANCA). U aktivnoj fazi bolesti često se mogu naći i specifična antitijela na kolagen tipa II te anti-matrilin-1 antitijela, iako su oni niske specifičnosti.
Slikovne metode. Ovisno o kliničkim manifestacijama (zahvaćenosti) bolesti, mogu se koristiti različite radiološke i/ili nuklearno-medicinske metode kojima se nastoji dokazati oštećenje hrskavičnoga tkiva (pregledni radiogram glave i pluća, kompjuterizirana tomografija i sl.). Danas se sve više govori i o ulozi PET/CT pretrage s fluorodeoksiglukozom u cilju, ne samo dijagnostike, nego i praćenja aktivnosti bolesti te odgovora na liječenje. Ehokardiografija i CT angiografija aorte koriste se za evaluaciju kardiovaskularnih simptoma, a CT ili MR glave s angografijom za evaluaciju radioloških simptoma.
Biopsija i histološki nalaz mogu pomoći u diferenciranju bolesti, ali ne predstavljaju osnovni alat u dijagnostici ovoga poremećaja.
|
Tablica 9.22. Predloženi dijagnostički kriteriji za RP (prema autorima i godini objave kriterija)
|
|
McAdam, 1976.
|
Postojanje najmanje tri od sljedećih nabrojanih obilježja: hondritis uške, neerozivni upalni poliartritis, nazalni hondritis, okularne manifestacije, hondritis hrskavica dišnoga sustava, audiovestibularno oštećenje. Patohistološka potvrda bolesti nije potrebna.
|
|
Damiani i Levine, 1979.
|
Najmanje jedan od tri navedenih McAdam kriterija uz histološku potvrdu bolesti ili najmanje dva McAdam kriterija uz pozitivan klinički odgovor na primjenu dapsona ili kortikosteroida
|
|
Michet, 1986.
|
Potvrđen hondritis dvije od tri lokalizacije (uho, nos, dišni putevi) ili potvrđen jedan hondritis (uho, nos, dišni put) uz druga dva manja kriterija (audiovestibularni poremećaj, okularne manifestacije, seronegativni artritis).
|
Liječenje. Primjena nesteroidnih protuupalnih lijekova čini temelj simptomatskoga liječenja ovih bolesnika. U liječenju bolesnika s blagim oblikom RP-a koristi se dapson (50 - 200 mg/dan per os) ili kolhicin (0,6 mg 2 – 4 x/dan), dok se kod bolesnika koji ne reagiraju na ove lijekove te kod bolesnika koji imaju teže kliničke manifestacije (zahvaćanje hrskavica dišnih puteva, vaskulitis i druge sistemske manifestacije) primjenjuju sistemski glukokortikoidi, najčešće peroralni prednizolon u dozi od 0,5 - 1 mg/kg/dan, a u najtežim oblicima opravdana je i pulsna terapija metilprednizolonom (500 - 1000 mg intravenski kroz tri dana). Kod bolesnika s učestalim recidivima ili onih koji ne reagiraju na primjenu glukokortikoida opravdana je primjena drugih imunosupresiva i imunomodulatora koji predstavljaju drugu liniju liječenja kao što su ciklofofamid (1 mg/kg/dan kroz dva tjedna uz povećanje doze za 25 mg svaka dva tjedna prema potrebi), azatioprin (2 mg/kg/dan), ciklosporin (5 mg/kg/dan) ili metotreksat (15 - 25 mg/tjedno). U trećoj liniji liječenja primjenjuju se TNF inhibitori (infliksimab, etanercept, adalimumab), rituksimab (monoklonalno antitijelo usmjereno na CD20+ koje dovodi do deplecije B-limfocita), anakinra (antagonist IL-1 receptora) , tocilizumab (antagonist IL-6 receptora) te abatacept (inhibitor koaktivacije T-limfocita).
Definicija i epidemiologija. IgG4 bolest predstavlja imunološki posredovanu bolest koju karakterizira tkivna infiltracija CD4+ T-limfocitima i IgG4+ plazma stanicama s posljedičnim fibroupalnim procesom koji rezultira stvaranjem brojnih fibroznih tumefakcija. Iako bolest može zahvatiti gotovo bilo koji organ i tkivo, najčešće su zahvaćene žlijezde slinovnice, gušterača, peritoneum i retroperitonealno vezivno tkivo. Radi se o relativno rijetkoj bolesti, iako je njezina stvarna učestalost podcijenjena s obzirom na novi klinički entitet o kojemu se priča unazad zadnjih dvadesetak godina. Bolest se uglavnom pojavljuje u dobi između petoga i sedmog desetljeća života s nešto češćom ukupnom pojavnošću kod muškaraca (gušterača je češće zahvaćena kod muškaraca, dok su žlijezde slinovnice češće zahvaćene kod žena).
Etiopatogeneza IgG4 bolesti nije poznata. S obzirom na prisutnu infiltraciju IgG4+ plazma stanicama u zahvaćenim tkivima i kliničko poboljšanje nakon primjene rituksimaba koji dovodi do deplecije B-limfocita, smatra se da oni imaju bitno mjestu u cijeloj patofiziologiji poremećaja (specifični imunološki odgovor na neki, za sada nepoznati antigen), no danas se sve više govori o primarnom poremećaju T-limfocita (CD4+) koji se također nalaze u upalnom infiltratu zahvaćenih tkiva. Također, smatra se da stvoreni IL-13 i TGF-β imaju glavnu ulogu u aktivaciji fibroblasta i nastanku fibroblastičnih tumefakcija.
Klinička slika. IgG4 bolest može zahvatiti bilo koji organ ili tkivo u tijelu pa stoga može imati širok raspon kliničkih prezentacija. Glavno kliničko obilježje podrazumijeva stvaranje fibroznih tumefakcija koje mogu rezultirati različitim poremećajima. Kod ovih bolesnika izraženiji su lokalni simptomi, a samo kod manjega broja nalazimo sistemske manifestacije kao što su vrućica, umor, malaksalost ili gubitak na tjelesnoj težini. Najčešće kliničke manifestacije su kronični pankreatitis, sijaloadenitis, tubulointersticijski nefritis, dakrioadenitis, peritonitis te periaortritis. Kod više od polovine bolesnika bolest se prezentira istodobnim zahvaćanjem dva ili više organa, a ako se radi o zahvaćanju samo jednoga organa, najčešće se radi o gušterači. Također, kod oko polovine bolesnika nađu se i atopijska oboljenja kao što su alergijski rinokonjuktivitis, dermatitis i astma.
Dijagnoza. Postavljanje dijagnoze često je otežano zbog širokih i nespecifičnih manifestacija koje mogu nalikovati na druga oboljenja kao što su tumori, sarkoidoza i sl., tako da je i sam proces dijagnostike usmjeren prema njima (slikovne metode, endoskopska obrada). Povišenu razinu IgG4 u serumu nalazimo kod većine bolesnika, ali ne kod svih, stoga se ovaj kriterij ne može uzeti kao temeljni ili jedini. Uz anamnestičke podatke (kliničku sliku), u dijagnostici je ključni patohistološki nalaz nađenih promjena koji uključuje gustu limfoplazmocitnu infiltraciju u kojemu dominiraju IgG4+ plazma stanice uz iregularnu fibrozu sa stvaranjem specifičnih tumoroznih promjena i obliterativni flebitis.
Diferencijalna dijagnoza IgG4 bolesti uključuje širok spektar bolesti i stanja: neoplastične bolesti (solidni tumori, limfomi), granulomatozne upalne (sarkoidoza, tuberkuloza, granulomatoza s poliangiitisom), primarni sklerozirajući kolangitis, retroperitonealna fibroza i sl.
Liječenje i prognoza. Kod većine bolesnika prati se spontana remisija bolesti. Kod izraženih kliničkih manifestacija u prvoj liniji liječenja koriste se kortikosteroidi (prednizon 0,6 mg/kg/dan) uz postupno smanjenje doze ovisno o kliničkom odgovoru. Kod bolesnika kod kojih se ne postiže zadovoljavajuća kontrola bolesti i klinički odgovor primjenjuje se rituksimab koji dovodi do deplecije B-limfocita ili neki od klasičnih imunomodulatora (mikofenolat mofetil, ciklofosfamid, azatioprin). Konačna prognoza bolesnika ponajprije ovisi o vremenu postavljanja dijagnoze i početka liječenja. Bolesnici koji se na vrijeme dijagnosticiraju i započnu liječiti imaju dobru prognozu i ne razvijaju kronične komplikacije koje su posljedica fibroznoga procesa.
Definicija. Osteoartritis (OA) je naziv za složen patološki proces obilježen oštećenjem zglobne hrskavice i periartikularnih struktura koji nastaje kao posljedica lokalnoga upalnog procesa u odgovoru na brojne genetske, metaboličke i biomehaničke utjecaje. Radi se o najčešćem obliku artritisa koji se dugo vremena smatrao degenerativnim procesom vezanim uz starenje zbog čega je i nosio naziv osteoartroza, no uviđanjem da lokalna upala ima ključno mjesto u patofiziološkom zbivanju, on je zamijenjen današnjim nazivom.
Epidemiologija. OA predstavlja najčešću bolest zglobova koja se smatra bolešću starenja, s obzirom na to da se kod više od 80 % osoba starijih od 60 godina mogu dokazati radiološki znakovi bolesti, ponajprije na velikim zglobovima koji podnose veliko opterećenje (kuk, koljeno), iako je ona kod većine bolesnika asimptomatska. Učestalost simptomatskoga OA-a raste s daljnjim porastom životne dobi i tako postaje jedan od najučestalijih bolnih sindroma kod starijih osoba. OA koljena razvije oko 45 %, a OA kuka oko 25 % ukupne populacije u nekom trenutku života. S obzirom na spolnu raspodjelu bolesti, OA se češće javlja kod žena u odnosu na muškarce, osobito nakon menopauze.
Podjela. U načelu razlikujemo dva osnovna oblika OA: primarni i sekundarni. Primarni ili idiopatski OA javlja se bez prethodnoga oštećenja zgloba, a može biti generaliziran (zahvaća tri ili više zgloba) ili lokaliziran (zahvaća do dva zgloba). Sekundarni OA nastaje kao posljedica ranije poznatoga oštećenja zglobne hrskavice (trauma, kirurški zahvat, infekcija, metabolički i endokrini poremećaji i sl.).
Etiopatogeneza. OA nastaje kao posljedica poremećaja fiziološke obnove hijaline hrskavice koja se nalazi između dva zgloba tijela te amortizira i umanjuje trenje između koštanih dijelova i odgovora na sile tlaka i vlaka koje djeluju na zglob. Ona se sastoji u najvećem dijelu od vode (80 %), dok ostatak (20 %) čini organska tvar. U organskoj tvari dominantnu ulogu imaju hondrociti koji stvaraju kolagenska vlakna tipa II koja se međusobno udružuju u kolagensku mrežu i odgovaraju na sile vlaka te proteoglikane (glukozaminoglikan, hondroitin sulfat, keratan sulfat) koji na sebe vežu hijaluronsku kiselinu te daju hrskavici elastičnu komponentu koja je odgovorna za apsorpciju sile tlaka. Tijekom cijeloga života u samoj hijalinoj hrskavici odvija se simultano proces razgradnje i stvaranja navedenih molekula, i oni su u dinamičkoj ravnoteži. Određeni čimbenici mogu dovesti do opterećenja zgloba i poremećaja ove ravnoteže u smislu pojačane razgradnje koju ne može pratiti odgovarajuće obnavljanje te dolazi do oštećenja hijaline hrskavice. Ono izaziva lokalnu upalnu reakciju kao fiziološki odgovor na oštećenje i to se danas smatra središnjim događajem u nastanku OA-a, budući da stvoreni proupalni medijatori promoviraju daljnje hrskavično oštećenje. Glavni upalni medijatori u OA-u su IL-1 i TNF-α. Oni djeluju na hondrocite tako da ih potiču na nekontroliranu produkciju matriks metaloproteinaza (MMP) i kolagenaza, enzima koji dovode do daljnje razgradnje kolegenskih vlakana i proteoglikana. Početno se učincima ovih medijatora suprostavljaju medijatori koji stimuliraju proces stvaranja nove hrskavice, kao što su PAI-1 (inhibitor aktivatora plazminogena), IGF-1 (inzulinski faktor rasta) te tkivni inhibitori MMP, ali perzistencijom zglobnoga opterećenja koje potiče upalnu reakciju oni postaju insuficijentni za zaustavljanje procesa razgradnje hrskavice. Gubitkom kolagenske mreže dolazi do dezintegracije i nestajanja hijaline hrskavice pri čemu se sužava zglobna pukotina i dolazi do izravnoga kontakta zglobnih tijela. S obzirom na to da je hijalina hrskavica avaskularna i aneuralna struktura, ovaj proces nije praćen bolovima. Gubitkom hrskavice i njezinoga amortizirajućeg djelovanja između zglobnih tijela dolazi do pojačanoga trenja i oštećenja koštanih struktura zglobnih tijela pri čemu nastaju brojne mikrofrakture, što je praćeno nastankom bolova zbog bogate živčane mreže periosta. Kost odgovora na novonastalo stanje subhondralnom sklerozom te rubnim bujanjem koštanoga tkiva (osteofiti) s ciljem proširenja površine opterećenja čime se nastoji stabilizirati zglob. U ovoj fazi OA postaje simptomatski, bolesnici se tuže na bolove, a dolazi i do poremećaja funkcije u smislu ograničenja i nemogućnosti izvođenja nekih pokreta ili je njihovo izvođenje praćeno jakim bolovima.
Rizični čimbenici u razvoju OA. Čimbenike koji podržavaju gore opisani proces nastanka OA možemo podijeliti u nekoliko skupina: starenje, biomehanički utjecaji, metabolički utjecaji i genetski utjecaji.
Biomehanički utjecaji. Mehaničko opterećenje zgloba jedan je od najznačajnijih rizičnih čimbenika u razviju OA-a. Trajno opterećenje (sile tlaka ili vlaka) dovodi do mehaničkoga oštećenja zglobne hrskavice koje rezultira pokretanjem lokalne upalne reakcije koja dovodi do daljnjih promjena i oštećenja. Povećano opterećenje zgloba može biti posljedica poremećenih odnosa unutar samoga zgloba (stanja nakon traume ili operacije, kongenitalne anomalije, nestabilnost zgloba), povećane tjelesne težine (pretilost je snažan rizični čimbenik u razvoju OA kuka i koljena) ili poremećene statike pri čemu se opterećuje jedna skupina zglobova.
Metaboličko-endokrini utjecaji. Različite metaboličke i endokrinološke bolesti mogu biti povezane s razvojem OA, kao što su hemokromatoza, Wilsonova bolest, akromegalija i metabolički sindrom. Pretilost, osim mehaničkoga opterećenja koje može dovesti do povećanoga rizika od razvoja OA kuka i koljena, povezana je i s povećanom učestalosti OA šake, što se ne može protumačiti povećanim opterećenjem zgloba zbog čega se u ovoj poveznici treba misliti i na druge mehanizme (pretilost kao izvor kronične upale niskoga intenziteta).
Genetski utjecaji. Činjenica da u unutar nekih obitelji nalazimo povećanu učestalost OA-a, kao i povećanu učestalost između blizanaca sugerira na povezanost OA-a i genetskih čimbenika. Dosadašnja istraživanja pokazala su povećanu pojavnost OA-a kod osoba kod kojih su identificirani polimorfizmi određenih gena koji se povezuju sa stvaranjem kolagena i razvojem koštanoga sustava (COL2A1, GDF5, PACE4, TRPV1).
Starenje. Kako je već navedeno, starenje je jedan od značajnijih čimbenika u razvoju OA-a. Starenjem dolazi do strukturalnih promjena hijaline hrskavice (gubitak hondrocita, oksidativni stres) koje je čine vulnerabilnom i sklonom oštećenju te razvoju lokalne upale.
Klinička slika. OA je u načelu sporoprogresivna bolest koja može zahvatiti različite zglobove u tijelu, iako se najčešće radi o malim zglobovima šaka, koljenu i kuku. Temeljno kliničko očitovanje OA-a je bolnost zahvaćenoga zgloba koju bolesnici različito opisuju (tupa, žareća, kidajuća), a koja je povezana i koja se pogoršava pri kretnjama i opterećenjima zgloba. Karakteristično je i periodično pogoršanje simptoma, često povezano s vremenskim prilikama tako da bolesnici u tijeku bolesti opisuju razdoblja kada su bolovi intenzivniji i učestaliji koja se smjenjuju s razdobljima kada su bolovi manjega intenziteta i rjeđe se javljaju. Gotovo se kod svih bolesnika zajedno s bolovima javlja i zakočenost zglobova koja se javlja nakon spavanja (jutarnja zakočenost) ili u mirovanju, a traje do 30 minuta i smanjuje se u pokretu. S vremenom bolesnici navode pojavu osjećaja krepitacija, odnosno struganja unutar zgloba. Navedene kliničke promjene dovode do ograničenja pokreta što dovodi do poremećaja funkcije zgloba, a u najtežim oblicima dolazi i do razvija kontraktura i deformiteta. Uz navedena očitovanja kod nekih se bolesnika mogu naći i nestabilnost zgloba, atrofija periartikularnih mišića, izražene palpatorne koštane izbočine (osteofiti) te izljev u zglobnu čahuru.
OA šake. Najčešće pogođeni zglobovi šaka su distalni interfalangealni (DIP), a rjeđe mogu biti zahvaćeni i proksimalni interfalangealni (PIP) te prvi karpometakarpalni (CMC) zglob. Karakteristično se kod ovih bolesnika pojavljuju bolni čvorići na posterolateralnim stranama DIP (Heberdenovi čvorići) i PIP (Bouchardovi čvorići) zglobova, a s napredovanjem bolesti dolazi i do potpunoga povećanja volumena zgloba uslijed stvaranja osteofita te razvoja deformiteta šake u obliku ulnarne devijacije. OA šaka razvija se kroz dulji vremenski period (1 do 5 godina) tijekom čega se prati postupno slabljenje funkcionalnoga statusa, iako kod jednoga dijela bolesnika u fazi razvijene bolesti prestaje daljnji gubitak funkcionalne sposobnosti šake te se simptomi mogu smiriti. OA šake češće se viđa kod žena nego kod muškaraca.
OA koljena. Kod većine bolesnika pojava OA koljena je obostrana i simetrična te pokazuje snažnu povezanost s pretilošću i ranijim traumatskim oštećenjima. Bol je najjače izražen u području prednjega i medijalnoga dijela koljenskoga zgloba, a tipično se pojačava prilikom hoda uz stepenice ili kod ustajanja iz čučećega ili sjedećeg položaja. Postupno dolazi do smanjenja opsega kretnji fleksije i ekstenzije te hipotrofije kvadricepsa što kod bolesnika stvara osjećaj nestabilnosti. Kod dominantne zahvaćenosti medijalnoga dijela koljenskoga zgloba nastaje varus deformacija, a kod dominante zahvaćenosti lateralnoga dijela nastaje valgus deformacija. Kod većine bolesnika mogu se naći i izljev te pojava Bakerove ciste u stražnjoj poplitealnoj jami.
OA kuka. Za razliku od OA koljena koji se obično javlja obostrano, OA kuka se kod najvećega broja oboljelih razvija samo na jednoj strani. Osnovno kliničko obilježje je bol koja je najjači u prednjem dijelu proksimalne trećine natkoljenice i može se širiti gore prema preponi, straga prema glutealnoj regiji te dolje prema koljenu. Bolnost ograničava pokretljivost, a najmanje je izražena u položaju lagane fleksije i unutarnje rotacije. Hipotrofija okolne muskulature (kvadriceps, glutealni mišići) dovodi do dodatne nestabilnosti zgloba.
OA kralješnice. Pored zahvaćanja navedenih zglobova, OA može zahvatiti i zglobove kralježnice, osobito u vratnom i slabinskom dijelu, što se uobičajeno naziva cervikalna ili lumbalna spondiloza. Osim lokaliziranih bolova zahvaćenoga područja, kod ovih bolesnika česte su i komplikacije vezane uz stvaranje osteofita koji mogu vršiti kompresiju živčanih korijena (radikulopatija) ili kralježničnu moždinu (mijelopatija) s posljedičnim neurološkim manifestacijama (senzomotorni deficiti).
Posebni klinički oblici OA. Rano nastali OA naziv je za pojavu OA-a prije 45. godine života, a obično se radi o monoartikularnom OA-u koji se javlja na podlozi ranijega oštećenja (trauma, operacija) zgloba. Erozivni OA također je rijetki oblik OA-a kojega karakterizira izrazita i progresivna destrukcija zglobne hrskavice i perihondralne kosti praćena izraženom simptomatolgijom, gubitkom funkcije i lošijim kliničkim ishodima (invalidnost). Erozivni OA najčešće se javlja u DIP i PIP zglobovima.
Dijagnoza OA postavlja se na temelju kliničke slike i nalaza, a potvrđuje se slikovnim radiološkim metodama. Laboratorijska dijagnostika nema veću ulogu u postavljanju dijagnoze OA-a. Klasično radiografsko snimanje predstavlja pretragu izbora u dijagnostici OA-a, a tipični nalazi uključuju suženje zglobne pukotine, subhondralnu sklerozaciju, nalaz postraničnih koštanih osteofita te osteoporotične promjene i cistične formacije u području subhondralne kosti. Magnetska rezonanca (MRI) i ultrazvuk (UZV) omogućavaju detaljniju analizu periartikularnih mekotkivnih struktura i same hrskavice, ali nemaju dokazanu prednost pred klasičnom radiografijom. Kod bolesnika s prisutnim izljevom u zglobnu čahuru indicirana je punkcija te biokemijska analiza dobivene tekućine što ima veći diferencijalno dijagnostički značaj u isključenju drugih zglobnih oboljenja (npr. infektivni artritis) nego u postavljanju same dijagnoze OA-a.
Liječenje. Osnovni ciljevi liječenja bolesnika s OA-om su simptomatsko olakšanje te održavanje funkcije zgloba. U liječenju se primjenjuju farmakološke i nefarmakološke mjere. Lijekovi koji se koriste u liječenju OA-a su oni koji imaju kombinirano protuupalno i analgetsko djelovanje, kao što su acetaminofen i nesteroidni protuupalni lijekovi, a nerijetko se primjenjuju i glukokortikoidi u obliku intraartikularnih injekcija. Nefarmakološke mjere uključuju mjere fizikalne terapije te u najtežim oblicima kirurško liječenje.
Mjere fizikalnoga liječenja. Svi bolesnici s OA-om trebali bi biti podvrgnuti mjerama fizikalnoga liječenja. One, osim što djeluju na smanjenja simptome, djeluju i na poboljšanje funkcionalnoga statusa (povećanje opsega kretnji zahvaćenoga zgloba, jačanje okolne muskulature). Lokalna primjena leda ili grijanje zahvaćenih zglobova u fazi akutizacije simptoma može imati analgetski učinak.
Farmakološke mjere liječenja. Nesteroidni protuupalni lijekovi ili nesteroidni antireumatici (NSAID ili NSAR ) najčešće su korišteni lijekovi u liječenju OA-a. Oni smanjuju proizvodnju prostaglandina inhibicijom enzima ciklooksigenaze koja posreduje njihovo stvaranje iz arahidonske kiseline. Na raspolaganju su nam neselektivni NSAID-i koji inhibiraju oba enzima ciklooksigenaze (COX-1 i COX-2) te su zbog toga povezani s povećanim rizikom od nastanka probavnih nuspojava, s obzirom na to da inhibiraju i stvaranje prostaglandina u želučanoj sluznici gdje oni imaju protektivni učinak te selektivni NSAID-i (etorikoksib, celekoksib) koji inhibiraju samo COX-2 te ne dovode do gastričkih smetnji, ali povećavaju rizik od nastupa kardiovaskularnih incidenata (iako se on opisuje i kod drugih NSAID-a). Rizik od krvarenja iz probavnoga sustava povezanoga s primjenom neselektivnih NSAID-a u općoj populaciji je relativno nizak (1:6000), a povećan je kod starijih osoba (> 70 godina), uz istodobno uzimanje glukokortikoida, antikoagulantnih i antitrombocitnih lijekova, kod bolesnika s ranijim gastričkim oboljenjima te kroničnih bolesnika. Svim bolesnicima koji se ubrajaju u neku od rizičnih skupina preporuča se provođenje gastroprotekcije inhibitorima protonske pumpe tijekom provođenja terapije NSAID-ima. Kod primjene NSAID-a (uključujući i celekoskib) treba imati na umu i mogućnost nastanka nefrotoksičnosti (intesticijski nefritis, nefrotski sindrom, prerenalna azotemija, pogoršanje hipertenzije), osobito kod bolesnika starijih od 60 godina, bolesnika koji imaju kroničnu bubrežnu bolest, kardiomiopata te kod istodobne primjene diuretika u većim dozama. Interferencija NSAID-a s trombocitima može dovesti do sklonosti produženju vremena krvarenja. Osim NSAID-a, u liječenju OA-a može se koristiti i acetaminofen (2 - 4 g/dan) ali on ima znatno slabiji učinak od NSAID-a.
Intraartikularna primjena kortikostroida. Lokalna primjena kortikosteroida izravnom aplikacijom u zglob primjenjuje se samo kod bolesnika kod kojih se kontrola simptoma ne može postići opisanim farmakološkim mjerama i fizikalnim liječenjem. Iako dovode do smanjenja bola, nema jasnih dokaza o dugoročnom učinku na tijek bolesti. Najčešće se primjenjuju depo preparati, kao što je triamkinolon i to u vremenskim razmacima od 6 do 12 mjeseci. Osim kortikosteroida, na ovaj se način mogu primjenjivati i hijaluronska kiselina te plazma bogata trombocitima (PRP, engl. plasma rich platelet).
Kirurško liječenje indicirano je u najtežim oblicima OA-a kod kojih se navedenim metodama više ne može postići odgovarajuća kontrola simptoma bolesti, a dovodi i do poboljšanja funkcionalnoga stanja. Najčešće se primjenjuju u liječenju OA-a koljena ili kuka (ugradnja totalne ili parcijalne endoproteze).
Prognoza. OA je progresivna bolest čiji se tijek može usporiti mjerama fizikalnoga i farmakološkog liječenja što može odgoditi potrebu za kirurškim liječenjem. Bolesnici koji se danas podvrgavaju kirurškom liječenju također u konačnici imaju dobre rezultate i mogu imati dobar funkcionalni status i kvalitetu života.
INFEKTIVNI ARTRITIS I KOŠTANE INFEKCIJE
Definicija. Infektivni ili septički artritis predstavlja infektivnu bolest zgloba koju najčešće uzrokuju bakterije, rjeđe drugi uzročnici kao što su virusi ili gljivice, dok je osteomijelitis naziv za infekciju koštanoga tkiva. I septički artritis i ostemijelitis predstavljaju ozbiljna infektivna oboljenja koja su često rezistentna na samo antibiotsko liječenje te često zahtijevaju i kirurške intervencije i mogu biti povezane s lošijim kliničkim ishodima i zaostajanjem trajnih funkcionalnih oštećenja lokomotornoga sustava.
SEPTIČKI ARTRITIS
Epidemiologija. Učestalost septičkoga artritisa u općoj populaciji kreće se između 2 do 10 nova slučaja na 100 000 stanovnika godišnje, dok se ona penje na 30 do 70 kod osoba s preegzistirajućim bolestima zglobova. Iako se može javiti u bilo kojoj životnoj dobi, češće se javlja kod starijih soba i ima nešto veću učestalost kod žena nego kod muškaraca.
Etiopatogeneza. Infekcija zgloba može nastati na nekoliko načina: 1) direktnom inokulacijom uzročnika u zglobnu šupljinu prilikom ozljede zgloba ili instumentalnih zahvata na zglobu, 2) širenjem per continuitatem iz okolnih izvora infekcije u koštanom ili periartikularnom mekom tkivu te 3) hematogenom diseminacijom iz udaljenih žarišta. Glavni rizični čimbenici koji pogoduju nastanku osteoartritisa su uznapredovala životna dob, preegzistirajuće bolesti zglobova (reumatoidni artritis, osteoartritis, umjetni zglob) te prisutnost kroničnih, imunokompromitirajućih bolesti (šećerna bolest, kronični alkoholizam, kožne ulceracije i infekcije, maligna oboljenja, kronične infekcije).
Mikrobiologija. Najčešći uzročnici septičkoga artritisa (60 - 90 %) su gram-pozitivne bakterije među kojima su najčešće Staphylococcus spp, Streptococcus spp, Enterococcus spp te Corynebacterium spp. Nešto rjeđe uzročnici mogu biti i gram-negativne bakterije (5 - 25 %), kao što su Salmonella spp, Pseudomonas aeruginosa, Escherichia coli, Klebsiella pneumoniae, Enterobacter spp i Haemophilus influenzae, vrlo rijetko se radi o anaerobima (Fusobacterium spp, Bacteroides fragilis) i atipičnim bakterijama (Mycoplasma spp).
Klinička slika. Ovisno o načinu nastanka, septički artritis može biti monoartikularni (npr. nakon ozljede ili operacije) ili poliartikularni (npr. septikemija i hematogeni rasap bakterija). Kod odraslih su najčešće zahvaćeni zglobovi donjih udova (najčešće koljeno). Kod djece dominiraju opći nad lokalnim simptomima, dok kod odraslih nalazimo obrnutu situaciju. U kliničkoj slici dominiraju opći (sistemski) i lokalni simptomi i znakovi. Opće kliničke manifestacije uključuju febrilno stanje, umor i malaksalost, algični sindrom te gubitak teka. Lokalni simptomi i znakovi koji upućuju na septički artritis su bolnost i otečenost zgloba koji je hiperemičan i topao na dodir s ograničenim i bolnim kretnjama.
Dijagnoza septičkoga artritisa postavlja se na temelju kliničke slike i nalaza, laboratorijskih nalaza, slikovnih metoda te u nekih slučajeva punkcije zgloba i analize dobivenoga sadržaja. U laboratorijskim nalazima obično nalazimo leukocitozu s neutrofilijom, povišene vrijednosti C-reaktivnoga proteina i prokalcitonina. Slikovne metode (klasični rendgenogram, CT, MRI) slabo su korisne u početnoj fazi bolesti kada se dominantno uočava povećana količina zglobne tekućine, odnosno izljev. Koštane erozije, sužavanje zglobnoga prostora te oštećenja periartikularnih struktura obično postaju detektibilna kroz nekoliko dana do nekoliko tjedana (ovisno o napredovanju bolesti). U dijagnostici septičkoga artritisa može se koristiti i scintigrafija radio-obilježenim leukocitima koji se pojačano nakupljaju na mjestu inficiranoga zgloba. Punkcija i analiza dobivenoga sadržaja indicirana je kod svih bolesnika sa sumnjom na septički artritis. Punktat je makroskopski zamućen (nekada se dobije i čisti gnoj), a citološkim pregledom nalazimo povećani broj leukocita (> 2000/mcL) uz dominaciju polimorfonukleara. Indicirana je i mikrobiološka analiza punktata, a kod sumnje na hematogeno nastali artritis potrebno je uzeti i hemokulture.
Liječenje. Osnovu liječenja čini parenteralna antibiotska terapija koja kada se primjenjuje inicijalno mora pokriti široki spektar bakterija (streptokoki, stafilokoki, gram-negativne bakterije), a kasnije se prilagođava nalazu antibiograma. Preporučeno je krenuti kombinacijom vankomicina (2 x 1 g) i cefalosporina treće generacije (ceftriakson 1 x 1 - 2 g, cefotaksim 3 x 1 - 2 g, ceftazidim 3 x 1 - 2 g). Ukupno antibiotsko liječenje trebalo bi trajati 4 do 6 tjedana. U slučaju slaboga odgovora na samo antibiotsko liječenje primjenjuju se mjere kirurškoga liječenja (drenaža, operativno liječenje). Početno se preporuča imobilizacija i mirovanje, no po smirivanju simptoma potrebno je što ranije započeti s kretnjama i mjerama fizikalne terapije.
Prognoza. Unatoč antibiotskom liječenju i kirurškim metodama, septički artritis i danas predstavlja ozbiljno oboljenje čija se smrtnost kreće i do 10 %. Starija životna dob smatra se glavnim rizičnim čimbenikom mortaliteta.
SPECIFIČNI OBLICI SEPTIČKOGA ARTRITISA
Gonokokni artritis. Diseminirana gonokokna infekcija razvija se kod 0,5 do 3 % bolesnika oboljelih od gonoreje, spolno prenosive bolesti uzrokovane gram-negativnom bakterijom Neisseria gonorrhoeae. Tijekom takve diseminacije bolesti, zglobovi su jedno od najčešće pogođenih tkiva. Smatra se da je učestalost gonokoknoga artritisa relativno visoka i da se kreće oko 130 slučaja na 100 000 stanovnika godišnje. Bolest se može prezentirati u dva oblika: gnojni artritis bez kožnih promjena ili kao trijas koji čini tendosinovitis, poliartralgije i kožne promjene u obliku makulopapuloznoga ili vezikularnog osipa (papule i vezikule ispunjene su gnojnim sadržajem). Zahvaćenost zglobova obično je asimetrična s jednakom pojavnošću i u malim i u velikim zglobovima. U dijagnostičkom postupku presudnu ulogu ima epidemiološki podatak o urogenitalnoj infekciji, a definitivno se postavlja nalazom gonokokne DNA (PCR metoda) iz uzorka sinovijalne tekućine, kožne lezije ili kulture iscjetka. Liječenje se sastoji do jednokratne oralne primjene azitromicina (1 g) te primjene cefalosporina treće generacije u dozama, kao i kod opisanoga septičkog artritisa. Primjena azitromicina dokazano poboljšava eradikaciju gonokoka, a pokriva i druge moguće konkomitantne spolne infekcije (Chalamydia spp). Liječenje treba provoditi minimalno dva tjedna, a ovisno o kliničkom nalazu i dulje.
Artritis zglobne endoproteze. Infekcija endoproteze zgloba predstavlja ozbiljan klinički problem koji predstavlja izazov u liječenju, a može se naći kod 0,3 do 1,7 % bolesnika nakon ugradnje endoproteze kuka i 0.8 do 1,9 % bolesnika nakon ugradnje endoproteze koljena. Rizik od nastanka ovih infekcija dva puta je viši kod bolesnika s reumatoidnim artritisom u odnosu na druge populacije bolesnika. Ovisno o vremenu pojavnosti, razlikujemo tri skupine infekcija endoproteza: 1) rana infekcija (unutar tri mjeseca od operativnoga zahvata), odgođena infekcija (infekcija u periodu od trećeg mjeseca do godine dana od operativnoga zahvata) te kasna infekcija (javlja se nakon više od godine dana od operativnoga zahvata). Rana i odgođena infekcija endoproteze obično nastaju kao posljedica kontaminacije tijekom samoga zahvata, dok je kasna infekcija obično posljedica hematogenoga širenja uzročnika iz nekoga drugog izvora. Najčešće se radi o stafilokoknim infekcijama, rjeđe o infekcijama gram-negativnim bakterijama. Kod manje od 10 % bolesnika kultura dobivene sinovijalne tekućine je negativna. Rizični čimbenici za razvoj ovih infekcija su infekcija kirurške rane, ranije kirurške manipulacije na zglobu, starija životna dob, reumatoidni artritis i druga kronična oboljenja (šećerna bolest, maligna bolest, alkoholizam). Klinička slika i dijagnostički postupak jednaki su kao i kod opisanoga septičkog artritisa. Liječenje provodi ortopedski tim, a osim prolongirane primjene antibiotika, u pravilu zahtijeva i uklanjanje infektivne endoproteze i kasniju reimplantaciju nove.
Virusni artritis. Artritis i artalgije često se nalaze kod bolesnika s virusnim infekcijama uzrokovanim hepatitis B i C virusom, citomegalovirusom, parvovirusom B19, rubeola virusom te virusom humane imunodeficijencije. Najčešće se radi o reaktivnoj promjeni, rjeđe o direktnoj sinovijalnoj infekciji. Klinička slika virusnih artritisa često oponaša reumatoidni artritis. Dijagnoza se postavlja nalazom pozitivnih specifičnih IgM protutijela u serumu bolesnika, a liječenje je kod većine bolesnika simptomatsko i bolest prolazi spontano kroz 4 do 6 tjedana.
Gljivični artritis obično se viđa kod imunokompromitiranih bolesnika. Najčešći uzročnici su Aspergillus spp, Candida spp, Cryptococcus spp i Nocardia spp. Dijagnoza se potvrđuje mikrobiološkom obradom sinovijalne tekućine, a liječenje ovisi o vrsti izolirane gljive.
OSTEOMIJELITIS
Uvod. Osteomijelitis označava infekciju kosti koja je najčešće uzrokovana bakterijama, rjeđe drugim mikroorganizmima, kao i septički artritis. Koštana infekcija može nastati hematogenim širenjem uzročnika iz udaljenoga žarišta, širenjem uzročnika per continuitatem iz okolnih struktura (koža, zglob) ili direktnom inokulacijom tijekom traume ili kirurškoga zahvata. Najčešći uzročnik je S. aureus, s obzirom na to da kolnizira kožu kod više od 30 do 40 % osoba u općoj populaciji. Najčešći izvori infekcije prilikom hematogenoga nastanka osteomijelitisa su celulitis, penumonije i infekcije urinarnoga trakta. Rizični čimbenici za razvoj osteomijelitisa jednaki su kao i za septički artritis.
Klinička slika. Glavne kliničke manifestacije osteomijelitisa obuhvaćaju ograničenu bolnost, otok i crvenilo kože iznad mjesta upale. Kod teških gnojnih infekcija mogući je i razvoj kutanih fistula s gnojnom sekrecijom. Kod jednoga dijela bolesnika prisutni su i sistemski znaci upale (vrućica, slabost, algični sindrom).
Dijagnoza se postavlja na temelju kliničke slike i kliničkoga nalaza, laboratorijskih nalaza koji slijede klasični obrazac za bakterijske infekcije (leukocitoza, neutrofilija, povišeni C-reaktivni protein) te slikovnih metoda među kojima je najosjetljivija magnetna rezonancija. U dijagnostici se može koristiti i scintigrafija radio-obilježenim leukocitima koji se pojačano nakupljaju na mjestu koštane infekcije.
Liječenje ostemijelitisa provode ortopedi, a sastoji se od parenteralne prolongirane primjene antibiotika koji pokrivaju široki spektar bakterija (slično kao i kod septičkoga artritisa) te kirurškoga zahvata.
ARTROPATIJE UZROKOVANE ODLAGANJEM KRISTALA
Artropatije uzrokovane odlaganjem kristala predstavljaju posebnu skupinu bolesti zglobova koje karakteriziraju oštećenja nastala odlaganjem različitih anorganskih kristala u području zglobnih struktura, a među njima je najznačajniji urični artritis ili giht. U ostale, rijetke oblike ubrajamo artropatiju uzrokovanu odlaganjem kalcijskoga pirofosfat-dihidrata te artropatiju uzrokovanu odlaganjem apatita (kalcijskoga fosfata).
URIČNI ARTRITIS (GIHT)
Definicija. Urični artritis (giht) naziv je za upalnu bolest zglobova koja nastaje kao posljedica poremećaja metabolizma mokraćne kiseline, odnosno hiperuricemije. Mokraćna kiselina nastaje kao konačni produkt katabolizma (razgradnje) purinskih baza, u tjelesnim tekućinama nalazi se u ioniziranom obliku (mononatrijev urat) te se izlučuje najvećim dijelom putem bubrega, a manjim dijelom putem probavnoga sustava. U stanjima prezasićenosti tjelesnih tekućina mokraćnom kiselinom (hiperuricemija; urati > 360 umol/L) ona može stvarati kristalne strukture koje se mogu odlagati u različita tkiva i organe među kojima su i zglobovi te uzrokovati njihova oštećenja. Više o fiziologiji i metabolizmu mokraćne kiseline opisano je u poglavlju Poremećaji metabolizma mokraćne kiseline.
Epidemiologija. Urični artritis predstavlja najčešću metaboličku artropatiju u kliničkoj praksi s prevalencijom koja se kreće od 2,5 do 5 % unutar opće populacije. Njegova pojavnost bilježi stalni porast što se povezuje sa sve češćom pojavnosti hiperuricemije uvjetovane promjenom prehrambenih navika (povećani unos purina životinjskim mesom i alkoholom), kao i produljenim životnim vijekom. Urični artritis češće se javlja kod muškaraca u odnosu na žene i to u omjeru 3 - 4:1, iako se taj omjer smanjuje u starijoj životnoj dobi (porast učestalosti hipeuricemije kod žena uslijed gubitka estrogenskoga urikozuričnog učinka).
Etiopatogeneza uričnoga artritisa izravno je povezana s hiperuricemijom, a njezini uzroci i mehanizmi nastanka opisani su u poglavlju Poremećaji metabolizma mokraćne kiseline. Formiranje kristala mononatrijeva urata (MSU) u zglobovima i mekim tkiva odvija se puno prije samih kliničkih manifestacija. Oni se najprije kristaliziraju u strukture koje nazivamo mikrotofi koji oblikuju rešetkaste strukture u površinskim dijelovima hijalne hrskavice, postupno rastu i ne stvaraju većih poteškoća. S vremenom dolazi do raspada rešetkaste strukture i rasipanja kristala koji aktiviraju sinovijalne makrofage koji ih potom fagocitiraju (vjeruje se da ovom rasapu pogoduju promjene pH ili temperature hrskavice). Aktivirani makrofazi počinju lučiti različite medijatore upale među kojima je najznačajniji interleukin-1 (IL-1) koji je odgovoran za nastanak lokalne upalne reakcije koja trajno perzistira i dovodi do oštećenja hrskavice, koštanih erozija i sinovijalne hipertrofije. Aktivacija sustava nespecifične imunosti od strane vlastitih antigena i posljedične upalne reakcije svrstava ovaj poremećaj u skupinu stečenih autoupalnih bolesti.
Klinička slika. Urični artritis može se klinički prezentirati u akutnoj i kroničnoj formi (Slika 9.8.). Klinički tijek bolesti je kroničan s povremenim akutizacijama odnosno egzacerbacijama. U konačnici dolazi do strukturnih oštećenja zgloba s posljedičnim deformitetima, kontrakturama i gubitkom funkcije.
Akutni urični artritis nastaje naglo, obično tijekom noći i nakon nagloga povećanja ili smanjenja razine mokraćne kiseline u serumu, što se najčešće viđa nakon pojačanoga unosa purina (pojačana konzumacija alkohola ili pojačan unos životinjskoga mesa) ili uslijed primjene lijekova koji interferiraju s metabolizmom mokraćne kiseline. Najčešće se bolest prezentira zahvaćanjem samo jednoga zgloba (iako nekada može i više njih) i to je kod najvećega broja bolesnika zglob nožnoga palca (prvi metatarzofalangealni zglob). Klasično se javljaju otok, bol, crvenilo i otežane kretnje. Kod određenoga broja bolesnika mogu biti prisutni i sistemski znaci upale (febrilno stanje). Tegobe bez liječenja traju oko deset dana, a potom se spontano povlače. S vremenom napadaji postaju sve jači i učestaliji. Osim navedenoga zgloba palca, akutni napadaj gihta može se odviti i na drugim zglobovima (s izuzetkom kukova i ramena koji su rijetko zahvaćeni), kao i na periartikularnim mekim tkivima, kao što je svod stopala.
Kronični urični artritis obilježava stavranje tofa, odnosno tumefakcija građenih od kristala mononatrijeva urata. Najprije se uočavaju u području kože u hrskavica ušnih školjki, a kasnije se uočavaju i na drugim mjestima oko zglobova gornjih i donjih ekstremiteta. Oni su bezbolni, postupno rastu, a u najtežim oblicima stvaraju i egzulceracije pri čemu dolazi do izlaska guste mase na površinu kože što može nalikovati na apsces i fistulu. Oni mogu vršiti i pritisak na okolne krvne žile i živce te dovesti do specifičnih neurocirkulacijskih ispada ili kompresivnih simptoma.
Slika 9.8. Kliničke manifestacije uričnoga artritisa: kronični urični artritis s tofima (lijevo), depoziti mononatrij-urata (sredina), tofi olekranona burze (desno)
Dijagnoza se postavlja na osnovi kliničke slike i nalaza, laboratorijskih nalaza i slikovnih metoda. Osnovni laboratorijski nalaz ovih bolesnika je hiperuricemija koja je prisutna u više od 95 % slučajeva, a u akutnom napadaju nalazimo i leukocitozu te povišeni C-reaktivni protein kao reaktant akutne faze. Slikovne metode, osobito klasični rendgenogram, u ranim fazama ne pokazuju patološke promjene, no s vremenom se mogu uočiti indirektni i direktni znaci hrskavičnoga i koštanoga oštećenja (suženje zglobne pukotine, koštane erozije i sl.). Primjena ultrazvuka korisna je u detekciji tofa. U nejasnim slučajevima punkcija i analiza zglobe tekućine može biti ključna u postavljanju dijagnoze, a temeljni nalaz uključuje identifikaciju kristala mononatrijeva urata svjetlosnom mikroskopijom.
Liječenje uričnoga artritisa sastoji se od: 1) liječenja akutnoga napadaja, 2) liječenja hiperuricemije te 3) profilakse ponovnih napadaja i progresije kroničnih oštećenja.
Liječenje akutnoga napadaja. U ovoj fazi osnovu liječenja čini suzbijanje upale, a ne hiperuricemije jer, kako je to već navedeno, naglo snižavanje vrijednosti serumskih urata može biti poticaj za nastanak i prolongiranje akutnoga napadaja. Lijekovi koje koristimo u ovoj fazi su nesteroidni protuupalni lijekovi, kolhicin, glukokortikoidi te inhibitori interleukina-1. Potrebno je započeti liječenje akutnoga napadaja što je ranije moguće (najbolje unutar 12 sati od simptoma atake).
Lijek izbora je kolhicin u dozi od 1 mg te potom nastaviti s dozom od 0.5 mg 1 sat nakon početne doze. Navedena shema može se ponoviti drugi i treći dan. Alternativa kolhicinu u liječenju akutnoga napadaja su NSAR-i ili glukokortikoidi kroz 3 do 5 dana.
Od NSAR-a se najčešće koriste naproksen (2 x 500 mg/dan) ili indometacin (3 x 25 - 50 mg/dan), a primjenjuju se sve dok ne dođe do simptomatskoga poboljšanja. Njihova primjena ne preporuča se kod bolesnika s ulkusnom bolesti te oslabljenom funkcijom bubrega. Kod rizičnih bolesnika potrebna je i gastroprotekcija inhibitorima protonske pumpe (životna dob > 70 godina, ranije gastričke smetenje, uzimanje lijekova koji djeluju na koagulacijski sustav, kronični bolesnici).
Glukokortikoidi su najučinkovitiji u suzbijanju simptoma akutnoga napadaja gihta, a imaju i najbrže djelovanje. Obično se primjenjuju kod bolesnika s kontraindikacijom za primjenu NSAR-a ili kolhicina (kao što su oštećena bubrežna funkcija). Oni se mogu primjenjivati intravenski (metilprednizolon 40 mg/dan) ili peroralno (prednizon 40 - 60 mg/dan) kroz tri do pet dana nakon čega se prekida bez prethodnoga snižavanja doze ili se mogu primjenjivati u navedenoj dozi, a potom se postupno snižava doza kroz 7 do 10 dana. Opravdana je i intraartikularna primjena kortikosteroida kod monoartikularnoga akutnog gihta koji slabo reagira na gore navedenu terapiju (triamkinolon 10 - 40 mg). Inhibitori IL-1 kao što su anakinra, kanakinumab i rilonacept primjenjuju se iznimno rijetko kod bolesnika kod kojih se ne može postići kontrola akutnoga gihta opisanim lijekovima (NSAR, kolhicin, kortikosteroidi).
Liječenje između napada. Liječenje bolesnika nakon (odnosno između) akutnoga napada gihta usmjereno je na liječenje hiperuricemije jer se time sprečavaju novi napadaji, ali i prevenira kronično oštećenje zglobova. Osnovu liječenja hiperuricemije čine nefarmakološke i farmakološke mjere.
Nefarmakološke mjere liječenja hiperuricemije podrazumijevaju smanjenje unosa alkohola (osobito piva) i mesa, s obzirom na to da su oni bogati purinima te izbjegavanje uzimanja lijekova koji utječu na metabolizam mokraćne kiseline i povećavaju njezinu serumsku razinu (salicilati, diuretici, levodopa).
Farmakološko liječenje hiperuricemije u prvom redu podrazumijeva primjenu inhibitora ksantin-oksidaze, ključnoga enzima u stvaranju mokraćne kiseline. Dva najvažnija lijeka iz ove skupine su alopurinol i febuksostat. Alopurinol se početo primjenjuje u dozi od 100 mg/dan (50 mg za bolesnike s uznapredovalom bubrežnom bolesti) te se postupno povisuje svakih 2 do 4 tjedana do postizanja ciljnih vrijednosti urata za što je najčešće potrebna doza od 300 mg/dan (3x100 mg), a maksimalna doza alopurinola iznosi 800 mg/dan. Preosjetljivost na alopurinol javlja se kod oko 2 % bolesnika, obično unutar prvih nekoliko mjeseci. Počinje kao osip praćen svrbežom, a može napredovati sve do toksične epidermalne nekroze. Hepatitis i vaskulitis rijetko se razviju. Sve bolesnike koji uzimaju alopurinol treba upozoriti da u slučaju pojave osipa odmah prestanu s njegovim uzimanjem. Febuksostat ne izaziva takvu preosjetljivost, a ne zahtijeva niti prilagodbu doze kod poremećene bubrežne funkcije. Primjenjuje se u jednoj dnevnoj dozi od 40 mg, a može se povisi na 80 mg/dan do maksimalno 120 mg/dan. Uz primjenu ovoga lijeka moguća je pojava hepatalne lezije, a neke studije pokazale su i veću stopu fatalnih kardiovaskularnih incidenata u odnosu na bolesnike koji su uzimali alopurinol.
Primjena kolhicina i urikozurika je rjeđa u kontroli hiperuricemije. Kolhicin je uspješan samo u liječenju blage hiperuricemije i kod bolesnika koji imaju rijetke akutne napadaje. Najčešća indikacija za primjenu kolhicina u ovoj indikaciji je nastavak liječenja ako je on korišten u suzbijanju akutnoga napadaja, a profilaktička doza iznosi 0,6 mg 1 – 2 x/dan. Urikozurici su lijekovi koji smanjuju vrijednosti mokraćne kiseline smanjujući njezinu reapsorpciju u tubulima, a u tu se svrhu mogu koristiti probenecid (0,5 mg/dan) te lesinurad (200 mg/dan). Oni se obično dodaju inhibitorima ksantin-oksidaze ako se ne postižu ciljne vrijednosti urata samo njihovom monoterapijom. Kontraindicirani su kod bolesnika s oštećenom bubrežnom funkcijom.
Kod bolesnika s teškim oblikom gihta, kod kojih se hiperuricemija ne može kontrolirati standardnom terapijom u punim dozama, u obzir dolazi uvođenje peglotikaze (biološka intravenska terapija - pegilirana urikaza).
Prognoza. Uz pridržavanje dijetetskih mjera i pravilne farmakoterapije kod većine bolesnika održava se period remisije i učestalost akutnih napadaja gihta. Loše kontrolirani bolesnici u konačnici razvijaju trajna oštećenja zglobova koja završavaju gubitkom njihove funkcije i određenim stupnjem invalidnosti.
Definicija i epidemiologija. Fibromialgija (FM) predstavlja idiopatski, kronični bolni sindrom koji difuzno zahvaća koštano-mišićni sustav s klasičnim očitovanjem u vidu bolnosti, osjetljivosti i ukočenosti mišića cijeloga tijela, osobito vrata i leđa. Radi se o jednoj od najčešćih dijagnoza u kliničkoj reumatološkoj praksi i može se naći kod 3 do 10 % osoba opće populacije. Češće se pojavljuje kod žena, obično između drugoga i šestoga desetljeća života. FM predstavlja i jedan od bitnih socioekonomskih problema budući da polovina ovih bolesnika u nekom trenutku razvije smanjenu radnu sposobnost.
Etiopatogeneza. Točan uzrok i mehanizam nastanka FM-a nije poznat. U središte patogeneze danas se stavlja poremećaj središnjega i perifernoga živčanog sustava u pogledu stvaranja i prijenosa bolnoga podražaja, u podlozi genetskih, okolišnih i psihosocijalnih utjecaja.
Poremećaj središnjega i perifernoga živčanog sustava u pogledu FM-a podrazumijeva pojačanu podražljivost i odgovor živčanoga sustava na bolni podražaj, kao i smanjen inhibitorni odgovor kralježnične moždine na osjet bola. Kod ovih bolesnika mogu se naći povećane koncentracije neurotransmitera koji pojačavaju osjet bola, kao što su supstanca P, glutamat te NGF faktor (engl. nerve growth factor), kao i smanjenje koncentracije serotonina i triptofana koji imaju suprotan učinak. Također, kod ovih bolesnika može se naći i poremećaj na razini sustava opioidnih receptora koji pokazuju smanjenu sposobnost vezanja endogenih opioida. Pojačana stimulacija i aktivacija glija stanica središnjega živčanoga sustava rezultira povećanim otpuštanjem citokina, dušikovoga oksida i prostaglandina koji stimuliraju i pojačavaju stanje hiperekscitibilnosti kralješnične moždine što dovodi do ukočenosti mišića.
Na genetsku podlogu bolesti upućuje činjenica se bolest pojavljuje češće unutar pojedinih obitelji i između bližih srodnika, a što se povezuje s polimorfizmima gena uključenih u metabolizam, transport te stvaranje i funkcioniranje receptora za serotinin i druge monoamine. Vanjski i psihosocijalni čimbenici koji se povezuju s nastankom FM-a su virusne infekcije (HCV, HIV), mehaničke i kemijske traume, nespavanje te emocije (nezadovoljstvo, stres).
Klinička slika. Osnovne karakteristike FM-a su difuzan mišićni bol i ukočenost, umor i poremećaji spavanja te kognitivne smetnje. Bol je dobro lokaliziran, zahvaća više skupina mišića, a najintenzivniji je u području vrata i donjega dijela leđa. Bolesnici ga opisuju kao osjećaj žarenja ili kao mukli i tup. Često je prisutna hiperalgezija (prekomjerna osjetljivost na bolni podražaj) i alodinija (pojava bola na bezbolan podražaj, npr. dodir). Bol se pojačava tijekom dana, a prisutna je i noću. Podražaji poput hladnoga vremena, fizičke i psihičke iscrpljenost dovode do pojačanja tegoba. Većina bolesnika navodi i probleme sa spavanjem u vidu otežanoga usnivanja i čestoga buđenja zbog čega imaju isprekidan san koji dovodi do nastanka osjeta umora i iscrpljenosti koji dodatno pojačavaju osjet bola. Česte su i kognitivne smetnje u vidu gubitka koncentracije, težeg pamćenja i zaboravljivosti. Također, kod ove skupine bolesnika češće su i druge psihosomatske senzacije u vidu lupanja srca, nedostatka zraka, trnaca, bolova u trbuhu i sl.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza FM-a postavlja se na temelju isključivanja drugih bolesti i stanja zbog čega se ovi bolesnici obično podvrgavaju širokoj dijagnostičkoj obradi. Za potrebe diferenciranja dijagnoze potrebno je isključiti reumatoidni artritis i druge autoimune bolesti vezivnoga tkiva, metaboličke miopatije, kronične infekcije (hepatitis C, borelioza), endokrinopatije (hipotireoza, adrenalna disfunkcija), sindrom kroničnoga umora te psihička oboljenja. Temeljna tri simptoma koja bi trebala upućivati na FM su bol, umor i nesanica. U postavljanju konačne dijagnoze koristimo se ACR-ovim kriterijima iz 2016. godine koji se temelje na tri kriterija: 1) rasprostranjenost bola (WPI, engl. widesparead pain index) te težini simptoma (SSS, engl. symptom severity scale); 2) dužini trajanja simptoma te 3) odsutnost druge bolesti kojom bi se mogli opisati simptomi. Rasprostranjenost bola tumači se WPI indeksom koji podrazumijeva postojanje 19 točaka bolnosti raspoređenih u pet regija (Tablica 9.23.), pri čemu bolesnik navodi gdje je u posljednjih tjedan dana osjetio bol, dok se težina simptoma tumači SS skalom koja podrazumijeva bodovanje (0 - 3) osjećaja umora, težine hodanja, kognitivnih smetnji i generalnih somatskih tegoba. Na FM ukazuje WPI ≥ 7 i SS ≥ 5 (ili WPI 3 - 6 i SS ≥ 9) uz prisutnost simptoma koji traju u kontinuitetu dulje od tri mjeseca i nepostojanje druge bolesti kojom bi se objasnili simptomi.
|
Tablica 9.23. WPI indeks u određivanju rasprostranjenosti bola (svaka stavka nosi jedan bod)
|
|
Lijeva gornja regija (1)
Lijeva donja čeljust
Lijevo rame
Lijeva nadlaktica
Lijeva podlaktica
|
Desna gornja regija (2)
Desna donja čeljust
Desno rame
Desna nadlaktica
Desna podlaktica
|
Aksijalna regija (5)
Vrat
Gornji dio leđa
Donji dio leđa
Prsni koš
Trbuh
|
|
Lijeva donja regija (3)
Lijevi kuk
Lijeva natkoljenica
Lijeva potkoljenica
|
Desna donja regija (4)
Desni kuk
Desna natkoljenica
Desna potkoljenica
|
Liječenje FM-a sastoji se od farmakoloških i nefarmakoloških mjera. U farmakološkom liječenju glavno mjesto imaju triciklički antidepresivi (amitriptilin), inhibitori ponovne pohrane serotonina i noradrenalina (fluoksetin, patoksetin) te antiepileptici (gabapentin, pregabalin). Svi navedeni lijekovi djeluju tako da između ostaloga inhibiraju i prijenos signala bola iz kralježnične moždine do mozga. Kod bolesnika koji ne reagiraju na navedene lijekove mogu se uključiti NSAID-i s ili bez tramadola (opioide treba izbjegavati u ovih bolesnika). Nefarmakološke mjere uključuju mjere fizikalne terapije, psihoterapiju te izbjegavanje stresa i čimbenika koji pogoduju pogoršanju simptoma.
Prognoza. Radi se o kroničnom poremećaju koji zahtijeva pridržavanje farmakoloških i nefarmakoloških mjera liječenja kako bi se osigurala kontrola simptoma.
KOMPLEKSNI REGIONALNI BOLNI SINDROM
Definicija. Kompleksni regionalni bolni sindrom (CRPS, engl. complex regional pain syndrome) predstavlja klinički sindrom kojega obilježava kroničan, kontinuiran bol koja zahvaća jednu regiju tijela s pratećim senzomotoričkim neurološkim deficitom i simptomima poremećaja autonomnoga živčanog sustava (bljedoća, crvenilo, znojenje). Ranije korišteni nazivi za ovaj poremećaj su kauzalgija, refleksna simpatička distrofija te Sudeckova distrofija.
Epidemiologija. Prava učestalost ovoga poremećaja nije utvrđena, a provedene studije pokazuju različite rezultate koji se kreću u rasponu od 5 do 25 novooboljelih bolesnika na 100 000 stanovnika godišnje. Poremećaj se javlja tri do četiri puta češće kod žena nego u muškaraca i to obično u srednjoj i starijoj životnoj dobi. Dvostruko češće su zahvaćeni gornji u odnosu na donje ekstremitete. Kod samo 5 do 10 % oboljelih poremećaj se razvija spontano, dok kod ostalih nalazimo prethodnu traumu ili kirurški zahvat u području zahvaćene regije.
Podjela. U načelu razlikujemo dva temeljna oblika: CRPS tip I i CRPS tip II. CRPS tip I javlja se spontano, bez prethodnoga neurološkog oštećenja, dok se CRPS tip II javlja nakon neurološkoga oštećenja (trauma, kirurški zahvat) i puno se češće susreće u kliničkoj praksi. Također, CRPS se dijeli i na akutnu i kroničnu formu. U akutnom obliku uz bol nalazimo i klasične znakove upale (edem, hiperemija, toplina), dok se u kroničnoj fazi uz bol javlja atrofija kože i mišića zahvaćene regije.
Etiopatogeneza. Točan uzrok i mehanizam nastanka CRPS-a mije poznat, ali se u centar patofizioloških zbivanja stavlja disfunkcija autonomnoga živčanog sustava, neurogena upala te poremećaj na razini središnjeg živčanoga sustava. Kako je pojavnost ovoga poremećaja snažno povezana s ranijim oštećenjima perifernoga živčanog sustava (trauma, kirurški zahvat), to se smatra jednim od najznačajnijih rizičnih čimbenika, iako nema presudnu ulogu s obzirom na to da se poremećaj može javiti i bez prethodnoga neurološkoga oštećenja. Do sada nije utvrđena povezanost između težine ozljede i nastanka ovoga poremećaja, što ukazuje na ulogu drugih mehanizama u nastanku ovoga poremećaja. Osim ozljede, kao rizični čimbenici navode se i emocionalna nestabilnost te psihička oboljenja (depresija, anksioznost, sindrom ovisnosti). Na autonomnu disfunkciju kao bitan čimbenik u razvoju CRPS-a ukazuje češća pojavnost CRPS-a kod osoba sa znacima povećane simpatičke aktivnosti (pojačano znojenje, sklonost vazokonstrikciji pri izlaganju hladnoći) te prisutnost kliničkih znakova autonomne disfunkcije (edem i hiperemija u akutnoj fazi, hladnoća i bljedilo u kroničnoj fazi). Vjeruje se da je autonomna disfunkcija razlog hiperalgeziji (pojačana osjetljivost na bol), a tome u prilog govori i smanjenje bolnosti nakon blokade simpatikusa. Poremećena autonomna funkcija rezultira i promjenom tonusa krvnih žila zbog čega se javljaju lokalni cirkulacijski poremećaji (hiperperfuzija u akutnoj fazi, hipoperfuzija u kroničnoj fazi). Opisanom patofiziološkom procesu pridonose i neurogena upala te poremećaji središnjega živčanog sustava s obzirom na to da se pojava mišićne slabosti ne može pripisati samo edemu ili atrofiji tkiva uslijed cirkulacijskih poremećaja, a isto su pokazale i različite neuroradiološke studije provedene kod ovih bolesnika (pojavnost razlike u aktiviranim kortikalnim područjima prilikom stimulacije zdrave i oboljele ruke).
Klinička slika. Bolesnici s CRPS-om mogu razviti tri skupine kliničkih manifestacija: 1) poremećaji osjeta, 2) poremećaji motorike te 3) poremećaji autonomnoga živčanog sustava. CRPS najčešće zahvaća gornje ekstremitete, a znatno rjeđe zahvaća neke druge dijelove tijela.
Poremećaji osjeta. Glavno kliničko obilježje CRPS-a su bol i ispadi senzacije. Oni karakteristično zahvaćaju jednu regiju tijela, ali nisu ograničeni na područje inervacije jednoga živca ili živčanoga korijena. Bolesnici bol opisuju kao dubok, oštar i žareći. Može biti prisutan u mirovanju, a češće je provociran fizičkim i psihičkim utjecajima (mehanički rad, promjene temperature, promjene raspoloženja). Ispadi senzacije najčešće se očituju kao hiperalgezija (pojačana osjetljivost na bolne podražaje), alodinija (izazivanje osjeta bola nebolnim podražajima, npr. dodir) te hipoestezija (smanjena osjetljivost kože na vanjske podražaje).
Poremećaji motorike. Također, kod većine ovih bolesnika može se razviti neki od motoričkih deficita u području zahvaćene regije: mišićne slabost, tremor, pojačani tetivni refleksi, distonija ili mioklonizmi.
Poremećaji autonomnoga živčanog sustava. Kako je već navedeno, znaci autonomne disfunkcije jedno su od glavnih obilježja CRPS-a. U akutnoj fazi javljaju se hiperemija, edem i hiperhidroza, dok se u kroničnoj fazi javlja bljedoća i hladnoća posljedično vazokonstrikciji i hipoperfuziji. U uznapredovaloj fazi javlja se atrofija mišića, trofičke kožne promjene te promjene noktiju i dlaka.
Dijagnoza CRPS-a postavlja se isključivo na temelju kliničke slike i kliničkoga nalaza s obzirom na to da do sada nije nađen niti jedan marker koji bi se mogao koristiti u dijagnostičke svrhe. U postavljanju dijagnoze koristimo se tzv. „Budimpeštanskim kriterijima“ koji obuhvaćaju četiri skupine kriterija koji moraju biti zadovoljeni da bi se postavila dijagnoza CRPS-a (Tablica 9.24).
|
Tablica 9.24. Budimpeštanski kriteriji u postavljanju dijagnoze CRPS-a
|
|
Prvi kriterij: kontinuiran bol nesrazmjeran bilo kojem poticajnom događaju.
|
|
Drugi kriterij: pojava barem jednoga simptoma u tri od četiri navedene kategorije:
v osjetna (hiperestezija, alodinija),
v vazomotorna (asimetrija tempetaure i/ili boje kože),
v sudomotorna (edem, promjena znojenja, asimetrija znojenja),
v motorna/trofička (smanjen opseg pokreta, motorički deficiti, trofičke promjene).
|
|
Treći kriterij: pojava barem jednoga znaka u vrijeme procjene u dvije ili više navedene kategorije:
v osjetna (dokaz o hiperalgeziji i/ili alodiniji),
v vazomotorna (dokaz o asimetriji temperature ili promjene boje kože),
v sudomotorna (dokaz edema i/li promjene znojenja i/ili asimetrije znojenja),
v motorna/tofička (dokaz motorne disfunkcije i/ili smanjenoga opsega pokreta i/ili trofičkih promjena).
|
|
Četvrti kriterij: ne postoji niti jedna druga dijagnoza koja bolje objašnjava kliničku sliku.
|
Liječenje. U liječenju CRPS-a koristimo se nefarmakološkim i farmakološkim mjerama. Nefarmakološke mjere uključuju edukaciju bolesnika o prirodi bolesti i potrebi izbjegavanja provocirajućih čimbenika te mjere fizikalne terapije koje imaju za cilj spriječiti razvoj atrofije i kontraktura zahvaćenih mišića. Rano farmakološko liječenje uključuje primjenu nesteroidnih protuupalnih lijekova (npr. naproksen 2 x 250 - 500 mg/dan), dok se u težim slučajevima primjenjuju i glukokortikoidi (prednizon u početnoj dozi od 30 - 60 mg/dan uz postupno snižavanje ovisno o kliničkom odgovoru). Uključivanje tricikličkih antidepresiva (amitriptilin, nortriptilin) ili antikonvulziva (gabapentin) može imati povoljan utjecaj na tijek bolesti. U najtežim oblicima u obzir dolazi i regionalna živčana blokada kao i primjena invazivnih procedura (stimulacija kralježnične moždine, simpatektomija).
Prognoza bolesnika s CRPS-om je varijabilna i ovisi o težini kliničke slike. U načelu uz liječenje i pridržavanje mjera izbjegavanja provocirajućim čimbenicima bolesnici mogu imati povoljan ishod bez razvoja kroničnih komplikacija (atrofija, deformacija) koji mogu narušiti kvalitetu života i radnu sposobnost. Zahvaćenost šaka ima lošiju prognozu od zahvaćenosti drugih tjelesnih regija.
Definicija. Amiloidoza predstavlja poremećaj metabolizma proteina kojega karakterizira nakupljanje patoloških, netopljivih fibrilarnih (vlaknastih) proteina koje nazivamo amiloidima u ekstracelularnom prostoru različitih tkiva i organa s njihovom posljedičnom destrukcijom i poremećajem funkcije. Amiloidi predstavljaju heterogenu skupinu proteina koje karakterizira fibrilarna (vlaknasta) građa, netopljivost i visoka otpornost na djelovanje proteinaza, zbog čega i dolazi do njihovoga nakupljanja u tkivima i organima. Poznato je više desetaka vrsta amiloida, no najčešće amiloidoza nastaje zbog patološkoga nakupljanja AL-amiloida, AA-amiloida, transtiretina te β2-mikroglobulina. Njihova pojačana produkcija može biti povezana s različitim bolestima i stanjima zbog čega amiloidoza u većini slučajeva predstavlja sekundarni (posljedični) poremećaj i zato dijagnostika i liječenje moraju biti usmjereni na osnovni poremećaj u podlozi.
Klasifikacija i etiopatogeneza. Amiloidoza se može dijeliti na osnovi više parametara: prema nastanku se dijeli na nasljednu i stečenu; prema raspodjeli nakupljanja, dijeli se na sistemsku i lokaliziranu; a prema vrsti amiloida koji se nakuplja, dijeli se na AL amiloiodzu (amiloidoza lakih lanaca), AA amiloiodzu (reaktivna amiloidoza), transtiretinsku amiloidozu (obiteljska amiloidoza) te druge rjeđe, specifične oblike.
AL amiloidoza karakterizirana je nakupljanjem AL amiloida koji se sastoji od lakih lanaca imunoglobulina kojeg luče imunosne stanice (plazma stanice) zbog čega se ovaj oblik naziva još i amiloioza lakih lanaca. Ona može biti primarna (nepoznatoga uzroka) ili sekundarna koju susrećemo u bolestima koje karakterizira proliferacija B-limfocita (multipli mijelom, Waldenstromova makroglobulinemija, ne-Hodgkinovi limfomi).
AA amiloidoza naziva se još i reaktivna amiloidoza s obzirom na to da je karakterizirana nakupljanjem AA amiloida koji se ubraja u skupinu proteina reaktanata akutne faze, a stvara se u jetri kao odgovor na stanje trajne stimulacije upalnim procesom. Prema tome, AA amiloidoza nastaje sekundarno kao posljedica kronične infekcije (tuberkuloza, osteomijelitis, bronhiektazije) ili kroničnih upalnih (autoimunih ili autoupalnih) bolesti (reumatoidni artritis, seronegativni spondiloartritisi, upalne bolesti crijeva i sl.).
Transtiretinska amiloidoza najčešći je primjer nasljednoga oblika amiloidoze. Transtiretin je serumski protein koji sudjeluje u prijenosu tiroksina i retinola, a kodira ga gen za transtiretin (TTR). Opisano je više od 100 mutacija ovoga gena koje su povezane s nastankom transtiretinske amiloidoze, a najčešća je Val30Met (zamjena valina metioninom na poziciji 30). Kao rezultat ove mutacije nastaje promijenjeni transtiretin koji poprima odlike amiloida te ima sklonost nakupljanju duže perifernih i autonomonih živaca zbog čega se ovaj oblik bolesti naziva autosomno-dominantna obiteljska amiloidozna polineuropatija.
Amiloidoza povezana s hemodijalizom jedan je od najčešćih oblika sekundarne amioloidoze, a razvija se kod bolesnika koji se liječe postupkom hemodijalize kroz dugi niz godina. Ona nastaje kao posljedica nakupljanja β2-mikroglobulina koji čini normalni dio molekule MHC-I razreda, a ne prolazi kroz dijalizatnu membranu zbog čega se nakuplja u serumu i odlaže u druga tkiva i organe.
Ostali oblici amiloidoze uključuju senilnu i endokrinu amiloidozu. Senilna amiloidoza povezana je sa starenjem i uznapredovalom životnom dobi (pojavljuje se nakon osmoga desetljeća života), a najčešće se očituje kao senilna srčana amiloidoza uslijed nakupljanja nemutiranoga transtiretina i senilna moždana amiloidoza uslijed nakupljanja β2-amiloidina. U određenim endokrinim bolestima praćenim pojačanim stvaranjem polipetidnih hormona kao posljedica se može razviti sekundarna amiloidoza (karcinomi štitnjače, neuroendokrini tumori).
Klinička slika. Amiloidoza se klinički može manifestirati kao lokalizirana ili kao sistemska bolest. Lokalizirana amiloidoza karakterizirana je nakupljanjem amiloida samo na jednom mjestu, zahvaćajući samo jedan organ, dok se sistemska amiloidoza prezentira istodobnim manifestacijama od strane zahvaćenosti više organa ili organskih sustava. Kod većine bolesnika nalazimo simptome i znakove osnovne bolesti koja se nalazi u podlozi te sistemske (opće) simptome kao što su umor, malaksalost ili gubitak na tjelesnoj težini. Amiloidoza se najčešće manifestira kao srčano popuštanje (infiltrativna/restriktivna kardiomiopatija), kronična bolest bubrega (nefrotski sindrom), senzomotorna periferna neuropatija ili asimptoatskim uvećanjima određenih organa i tkiva (hepatomegalija, splenomegalija, makroglosija, periorbitalna purpura).
Dijagnoza. Postavljanje dijagnoze amiloidoze često je otežano i zakašnjelo zbog širokih i nespecifičnih kliničkih manifestacija. Ključni korak u ovom postupku je postaviti sumnju na amiloidozu, što obično proizlazi iz kliničke slike te patoloških nalaza dobivenih drugim dijagnostičkim metodama (npr. ehokardiografija, ultrazvuk bubrega i sl.). Potvrdu dijagnoze omogućava jedino patohistološka dijagnostika. Kod sumnje na sistemsku amiloidozu indicirana je aspiracija potkožnoga masnog tkiva trbuha gdje se kod najvećega broja bolesnika mogu dokazati agregati amiloida, a ako je ona negativna, može se pristupiti ciljanoj biopsiji zahvaćenoga tkiva ili organa (npr. biopsija bubrega ili endomiokardijalna biopsija). Kod sumnje na lokalizirani oblik amiloidoze indicirana je ciljana biopsija zahvaćenoga tkiva. Kod svih bolesnika s potvrđenom amiloidozom potrebno je usmjeriti daljnji dijagnostički put prema otkrivanju osnovne, uzročne bolesti (imunološka i hematološka obrada). Ako se ona provedenom obradom ne utvrdi, radi se o primarnoj amiloidozi.
Liječenje amiloidoze u prvom redu ovisi o etiologiji: hematološki poremećaj, imunološki poremećaj, kronična infekcija i dr. U slučaju primarne AL amiloidoze liječenje koje se provodi slično je onome u liječenju multiploga mijeloma. kod bolesnika dobroga općeg statusa preporuča se kemoterapija visokim dozama melfalana i deksametazona nakon čega slijedi transplantacija krvotvornih matičnih stanica. Osim toga, u liječenju se mogu koristiti i ciklofosfamid, lenalidomid, bortezomib te daratumumab. U razvoju su i lijekovi u obliku specifičnih protutijela koja se mogu vezati za istaloženi amiloid te dovesti do njegove degradacije. Uz ove, provode se i druge potporne metode liječenja ovisno o zahvaćenom organu.
Prognoza sekundarne amiloidoze povezana je s uzročnom bolesti i njezinim karakteristikama i mogućnostima liječenja. Primana amiloidoza obilježena je progresivnim tijekom i ako ju se ne prepozna i ne liječi, vrlo brzo završava smrtnim ishodom (primarna amiloidoza koja zahvaća srce i autonomna živčana vlakana bez liječenja imaju srednje preživljenje od 3 do 9 mjeseci). Kod bolesnika koji su podvrgnuti kemoterapiji i transplantacijskom liječenju medijan preživljenja kreće se oko pet godina.
- Adkinson NF Jr, Hamilton RG. Clinical history-driven diagnosis of allergic diseases: utilizing in vitro IgE testing. J Allergy Clin Immunol Pract. 2015 Nov–Dec:3(6):871–6.
- Adler RS. Imaging studies in the rheumatic diseases. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1723-30.
- Akin C. Mastocytosis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1706-1710.
- Anić B. Sustavna skleroza (sklerodermija). U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1390-2.
- Anić B., Babić-Naglić Đ. Reumatoidni artritis. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1371-8.
- Anić B., Babić-Naglić Đ. Seronegativni spondiloartritisi. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1380-6.
- Anić B., Bosnić D. Bolesti koje posreduju imunokompleksi. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1361-5.
- Anić B., Bosnić D. Sustavni eritemski lupus. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1386-90.
- Arachchillage DRJ, Laffan M. Pathogenesis and management of antiphospholipid syndrome. Br J Haematol. 2017 Jul;178(2):181–95.
- Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019;71(9):1400-12.
- Atkinson JP. Complement system in disease. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 240-6.
- Bardin T et al. Lesinurad in combination with allopurinol: a randomised, double-blind, placebo-controlled study in patients with gout with inadequate response to standard of care (the multinational CLEAR 2 study). Ann Rheum Dis. 2017 May;76(5):811–20.
- Bennett RM. Fibromyalgia, chronic fatigue syndrom and myofascial pain. U: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders;2016. p. 1817-23.
- Blažeković I, Bilić E, Žagar M, Anić B. Kompleksni regionalni bolni sindrom. Liječ Vjesn 2015;137:297–306.
- Block JA, Scanzello C. Osteoarthritis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1744-9.
- Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015 Nov;136(5):1186–205.
- Bosnić D., Mayer M. Dermatomiozitis i polimiozitis. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1393-5.
- Bossuyt X, Cohen Tervaert JW, Arimura Y, Blockmans D, Flores-Suárez LF et al. Position paper: Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat Rev Rheumatol. 2017 Nov;13(11):683–92.
- Bulur I, Onder M. Behçet disease: new aspects. Clin Dermatol. 2017 Sep–Oct;35(5):421–34.
- Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA. 2016 Jun 14;315(22):2442–58.
- Bykerk VP, Crow MK. Approach to the patient with rheumatic disease. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1712-8.
- Castañeda S, Blanco R, González-Gay MA. Adult-onset Still’s disease: advances in the treatment. Best Pract Res Clin Rheumatol. 2016 Apr; 30(2):222–38.
- Clauw DJ. Fibromyalgia and related conditions. Mayo Clin Proc. 2015 May;90(5):680–92.
- Craft J. The adaptive immune system. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 220-6.
- Crow MK. Systemic lupus erythematosus. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. , Philadelphia: Elsevier Saunders; 2016. p. 1769-77.
- Crow MK. The innate immune system. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 216-20.
- Cunningham-Rundles C. Primary immunodeficiency disease. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1677-87.
- Čikeš N. Poremećaji sustava komplementa. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1352-4.
- Čikeš N. Sindromi preklapanja bolesti vezivnog tkiva i nediferencirana bolest vezivnog tkiva. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1395-6.
- Čikeš N., Anić B. Klasifikacija reumatskih bolesti. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1369-71.
- Čikeš N., Morović-Vergles J. Sindromi vaskulitisa. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1396-403.
- Ćurković B. Osteoartritis, križobolja i vratobolja. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1409-15.
- Ćurković B. Procjena aktivnosti bolesti i procjena ishoda u reumatoidnom artritisu. Fiz. rehabil. med. 2007; 21(1-2):17-22.
- Dreskin SC. Urticaria and angioedema. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1693-8.
- Edwards NL. Crystal deposition diseases. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1811-6.
- Erkan D, Lockshin MD. What is antiphospholipid syndrome? Curr Rheumatol Rep 2004; 6:451.
- Fanouriakis A, Kostopoulou M, Alunno A, et al2019 update of the EULAR recommendations for the management of systemic lupus erythematosusAnnals of the Rheumatic Diseases 2019;78:736-745.
- Frković M., Jelušić M. Novosti u etiopatogenezi i liječenju IgA vaskulitisa. Paediatr Croat. 2017; 61 (Supl 1): 43-47.
- Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. 2016 Nov;28 (6): 636–44.
- Gagro A. Autoimunost i imunodeficijencije. Reumatizam 2016;63(Suppl 1):66–72.
- Gagro A. Autoinflamatorne bolesti: od klasiikacije do prognoze. Hrvatska proljetna pedijatrijska škola, XXXVI. seminar. Split, 2019.
- Gašparić I., Gašparić S., Gotovac N., Šantak G. Gigantocelularni arteritis vertebralnih arterija kao uzrok moždanog udara. Med Jad 2015;45(3-4):135-138.
- Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A et al; European Society for Immunodeficiencies. Clinical and Experimental Immunology. 2009:157(Suppl. 1):3–11
- Gnjidić Z. Reaktivni artritis. Medicus. 2012; 21(1):135-41.
- Goodman SM, Springer B, Guyatt G, Abdel MP, Dasa V, George M et al. 2017 American College of Rheumatology/ American Association of Hip and Knee Surgeons guideline for the perioperative management of antirheumatic medication in patients with rheumatic diseases undergoing elective total hip or total knee arthroplasty. Arthritis Rheumatol. 2017 Aug;69(8):1538–51.
- Gossec L, Baraliakos X, Kerschbaumer A et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 updateAnnals of the Rheumatic Diseases 2020;79:700-712.
- Grazio S, Novak S, Laktašić-Žerjavić N, Anić B, Babić-Naglić Đ, Grubišić F i sur. Prijedlog preporuka Hrvatskoga Reumatološkog Društva za liječenje odraslih bolesnika s aksijalnim spondiloartritisom i psorijatičnim artritisom biološkim lijekovima i ciljanim sintetskim molekulama, 2017. Reumatizam 2017;64(2):71–87.
- Green berg SA. Inflammatory myopathies. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1789-93.
- Hashem H, Kelly SJ, Ganson NJ, Hershfield MS.. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep. 2017 Oct 5;19(11):70.
- Hatemi G, Christensen R, Bang D, Bodaghi B, Celik AF, Fortune F et al.2018 update of the EULAR recommendations for the management of Behçet’s syndromeAnnals of the Rheumatic Diseases 2018;77:808-18.
- Heineke MH, Ballering AV, Jamin A, Ben Mkaddem S, Monteiro RC, Van Egmond M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schönlein purpura). Autoimmun Rev. 2017 Dec;16(12):1246–53.
- Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitisAnnals of the Rheumatic Diseases 2020;79:19-30.
- Inman RD. The spondyloarthropathies. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1762-9.
- Jelušić M., Frković M. Novi klasifikacijski kriteriji sistemskog eritematoznog lupusa, sindroma aktivacije makrofaga i vaskulitisa. Paediatr Croat. 2017; 61(Supl 1):25-32.
- Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015 Apr 11; 385(9976):1460–71.
- Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am. 2015 Feb;35(1):61–76.
- Kingdon J,Roscamp J, Sangle S, D´ Cruz D. Relapsing polychondritis: a clinical review for rheumatologists. Rheumatology (Oxford). 2018;57(9):1525-35.
- Knight CL, Nelson-Piercy C. Management of systemic lupus erythematosus during pregnancy: challenges and solutions. Open Access Rheumatol. 2017 Mar 10;9:37-53.
- Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y et al; EUSTAR Coauthors. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017 Aug;76(8):1327–39.
- Krmpotić D, Čikeš N, Krmpotić P. Paraneoplastični sindrom povezan s antifosfolipidnim protutijelima. Liječ Vjesn. 2004;126:5-6
- Kuljiš D, Čulič S, Armanda V, Sršen S. Mastocitoze u djece. Med Jad 2009;39(3-4):83-8.
- Laktašić-Žerjavić N. Recidivirajući polihondritis. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1408-9.
- Laktašić-Žerjavić N, Ćurković B. Artropatije zbog odlaganja kristala. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1417-20.
- Malaty HM, Lo GH, Hou JK. Characterization and prevalence of spondyloarthritis and peripheral arthritis among patients with inflammatory bowel disease. Clin Exp Gastroenterol. 2017 Sep; 10:259–63.
- Mandal J, Chung SA. Primary angiitis of the central nervous system. Rheum Dis Clin North Am. 2017 Nov;43(4):503–18.
- Mariette X. Sjogren syndrome. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1785-9.
- Markeljević J. Autoimunost i autoimune bolesti. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1359-61.
- Markeljević J. Sjogrenov sindrom. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1392-3.
- Martinović Kalitrena D. Sistemska skleroza: pregled. Reumatizam 2017;64(Suppl 1):1–7.
- Martinović Kalitrena D., Božić I. Fibromialgija: je li to reumatska bolest? Reumatizam 2016;63(Suppl 1):27–30.
- Matteson EL, Osmon DR. Infectios of bursae, joints and bones. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1805-10.
- Maurer M, Magerl M, Ansotegui I, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy. 2018;73(8):1575-96.
- Mayer M., Laktašić-Žerjavić N. Infekcijski artritis. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak, 2008. str. 1420-2.
- McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M et al. Effect of intra-articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: a randomized clinical trial. JAMA. 2017 May 16;317(19):1967–75.
- Milas-Ahić J. Novosti u liječenju uričnog artritisa. Medix. 2014;(113/114):113-7.
- Milas-Ahić J, Prus V, Šustić N, Višević R, Kovačević I, Kardum Ž. Krioglobulinemični vaskulitis kao manifestacija paraneoplastičnog sindroma - prikaz bolesnika. Reumatizam 2015; 62(1):27-30.
- Milavec-Puretić V. Problem dijagnoze i liječenja kronične idiopatske urtikarije. Medicus. 2007;16(1):27-31.
- Milone M. Diagnosis and management of immune-mediated myopathies. Mayo Clin Proc. 2017 May;92(5):826–37.
- Mitrović J, Morović-Vergles J, Martinović Kaliterna D, Anić B, Babić-Naglić Đ, Grazio S i sur. Prijedlog preporuka Hrvatskoga Reumatološkog Društva za liječenje bolesnika s reumatoidnim artritisom biološkim i ciljanim sintetskim lijekovima, 2017; Reumatizam 2017;64(2):65–70.
- Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295.
- Morović-Vergles J. Amiloidoza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak, 2008. str. 1407-8.
- Morović-Vergles J., Markeljević J. Imunodeficijencije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak, 2008. str. 1349-52.
- Dell JR. Rheumatoid arthritis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1754-62.
- Padjen I., Cerovac M., Mayer M., Anić B. Sistemski eritemski lupus: opis i kvantifikacija fenotipa bolesti. Reumatizam 2017;64(1):1–9.
- Pisetsky DS. Laboratory testing in the rheumatic diseases. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p.1718-23.
- Ramos-Casals M, Brito-Zerón P, Bombardieri S On behalf of the EULAR-Sjögren Syndrome Task Force Group, et alEULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapiesAnnals of the Rheumatic Diseases 2020;79:3-18.
- Registry Working Party. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014 Jul;134(1):116–26.
- Richette P, Doherty M, Pascual E, Barskova V, Becce F, Castañeda-Sanabria J et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis 2017;76:29–42.
- Salar O, Baker B, Kurien T, Taylor A, Moran C. Septic arthritis in the era of immunosuppressive treatments. Ann R Coll Surg Engl. 2014 Mar;96(2):e11–2.
- Saraux A, Pers JO, Devauchelle-Pensec V. Treatment of primary Sjögren syndrome. Nat Rev Rheumatol. 2016 Aug;1 (8):456–71.
- Schemoul J, Poulain C, Claudepierre P. Treatment strategies for psoriatic arthritis. Joint Bone Spine. 2018 Oct;85(5):537-44.
- Schieir O, Tosevski C, Glazier RH, et alIncident myocardial infarction associated with major types of arthritis in the general population: a systematic review and meta-analysisAnnals of the Rheumatic Diseases 2017;76:1396-404.
- Schunrrer-Luke Vrbanić T, Rubnić D. Osteoartritis. Medicina 2003;40:108-10.
- Schwartz LB. Systemic anaphylaxis, food allergy and insect sting allergy. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p.1698-703.
- Selmanović V, Dizdarević-Omerčahić A, Čengić A. Obiteljska mediteranska groznica: interesentni i izazovni prototip autoinflamatorne bolesti. Paediatr Croat. 2016; 60 (Supl 1):90-9.
- Siegel RM, Kastner DL. The systemic autoinflammatory diseases. U: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1739-44.
- Singer O, McCune WJ. Update on maintenance therapy for granulomatosis with polyangiitis and microscopic polyangiitis. Curr Opin Rheumatol. 2017 May;29(3):248–53.
- Singh JA, Cameron C, Noorbaloochi S, Cullis T, Tucker M, Christensen R et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta-analysis. Lancet. 2015 Jul 18;386(9990):258–65.
- Smolen JS, Landewé RBM, Bijlsma JWJ, et al EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update Annals of the Rheumatic Diseases 2020;79:685-99.
- Spiera RF. Polymyalgia rhematica and temporal arteritis. U: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1801-5.
- Stipić-Marković A., Čvorišćec B. Alergijske bolesti i pseudoalergijske reakcije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak, 2008. str. 1355-9.
- Stone JH. The systemic vasculitides. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1793-801.
- Tajerian M, Clark JD. New concepts in complex regional pain syndrome. Hand Clin. 2016 Feb;32(1):41–9.
- Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. 2016 Jun 30;374(26):2563–74.
- Tektonidou MG, Andreoli L, Limper M et al. EULAR recommendations for the management of antiphospholipid syndrome in adultsAnnals of the Rheumatic Diseases 2019;78:1296-304.
- Thong B, Olsen NJ. Systemic lupus erythematosus diagnosis and management. Rheumatology (Oxford). 2017 Apr 1;56(Suppl 1): i3–13
- van Durme CM, Wechalekar MD, Buchbinder R, Schlesinger N, van der Heijde D, Landewé RB. Nonsteroidal anti-inflammatory drugs for treatment of acute gout. JAMA. 2015 Jun 9;313(22):2276–7.
- Varga J. Systemic sclerosis (sclerodermia). In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1777-85.
- Wasserman SI. Approach to the patinet with allergic or imunologic disease. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p.1674-7.
- Woodworth TG, Suliman YA, Li W, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016 Nov;12(11):678–91.